Blenrep
aef7c34c-fef8-407c-99c0-a68aded53c60
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
BLENREP is indicated in combination with bortezomib and dexamethasone for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least two prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent.
Dosage and Administration
• The recommended dosage of BLENREP, in combination with bortezomib and dexamethasone, is 2.5 mg/kg as an intravenous infusion over 30 minutes once every 3 weeks for 8 cycles, followed by BLENREP 2.5 mg/kg every 3 weeks as a single agent. ( 2.2 ) • See Full Prescribing Information for instructions on preparation and administration. ( 2.4 )
Contraindications
None.
Warnings and Precautions
• Thrombocytopenia: Monitor complete blood counts at baseline and periodically during treatment. Withhold or reduce the dosage based on severity. ( 2.3 , 5.3 ) • Embryo‑fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to fetus and to use effective contraception. ( 5.4 , 8.1 , 8.3 )
Adverse Reactions
In the BVd arm of the clinical study, 98% of patients required a dosage modification for any component of treatment for an adverse reaction, including 87% who required a dosage modification of BLENREP [see Clinical Studies ( 14 )] . Eighty-three percent of patients required a dosage modification of BLENREP for ocular toxicity based on ophthalmic exam findings or other ocular adverse reactions as defined by the Common Terminology Criteria for Adverse Events (CTCAE) [see Adverse Reactions ( 6.1 )] . There were high rates of dosage modifications in early treatment cycles. By Cycle 3, 53% of patients had a dosage interruption or reduction, 7% had discontinued treatment, and only 40% received the planned dose of BLENREP. The recommended dosage modifications for ocular toxicity based on ophthalmic exam findings are provided in Tables 1 and 2. Ophthalmic exam findings include both corneal exam findings and change in BCVA as assessed by an eye care professional. The overall grade of ophthalmic exam findings is based on the worst finding in the worst affected eye, based on either corneal exam finding or a change in BCVA. Corneal exam findings may or may not be accompanied by changes in BCVA or ocular symptoms. • Do not re‑escalate the dose of BLENREP after a dosage reduction is made for ocular toxicity based on ophthalmic exam findings. • In the clinical study, 67% of patients required a dosage interruption of BLENREP for ocular toxicity that lasted longer than 3 weeks (time between doses, median: 5.7 weeks [range: 3 to 31 weeks]). The recommended dosage modifications for other adverse reactions, including dosage modifications for ocular adverse reactions based on the CTCAE, are provided in Table 3 . Table 1. Recommended Dosage Reductions of BLENREP for Adverse Reactions a Reduced Dosage Level 2 is specific to dosage reductions due to ocular toxicity based on ophthalmic exam findings. BLENREP Reduced Dosage Level 1 1.9 mg/kg every 3 weeks Reduced Dosage Level 2 a 1.9 mg/kg every 8 weeks Table 2. Recommended Dosage Modifications for Ocular Toxicity Based on Ophthalmic Exam Findings a BCVA = best‑corrected visual acuity. a Mild superficial keratopathy (documented worsening from baseline). Refer to Table 3 for recommended dosage modifications for other ocular adverse reactions. b Microcyst‑like deposits are considered at least a Grade 2 finding. Withhold BLENREP if any microcyst‑like deposits are observed. Severity Ophthalmic Exam Findings [see Warnings and Precautions ( 5.1 )] Recommended Dosage Modification Grade 1 Corneal Exam Findings: Mild superficial punctate keratopathy a and/or Change in BCVA: Decline from baseline of 1 line on Snellen Equivalent BCVA Continue treatment at current dosage. Grade 2 Corneal Exam Findings: Moderate superficial punctate keratopathy, patchy microcyst‑like deposits b , peripheral sub‑epithelial haze, or a new peripheral stromal opacity and/or Change in BCVA: Decline from baseline of 2 lines on Snellen Equivalent BCVA and not worse than 20/200 Withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less. Resume treatment at Reduced Dosage Level 1 as per Table 1 . If recurrent Grade 2 or 3 ocular toxicity is experienced, resume treatment at Reduced Dosage Level 2. Grade 3 Corneal Exam Findings: Severe superficial punctate keratopathy, diffuse microcyst‑like deposits b involving the central cornea, central sub‑epithelial haze, or a new central stromal opacity and/or Change in BCVA: Decline from baseline of 3 or more lines on Snellen Equivalent BCVA and not worse than 20/200 Grade 4 Corneal Exam Findings: Corneal epithelial defect or corneal ulcer, with or without infection and/or Change in BCVA: Decline to Snellen Equivalent BCVA of worse than 20/200 Consider permanent discontinuation of BLENREP. If continuing treatment, withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less. For patients previously on 2.5 mg/kg every 3 weeks, resume treatment at Reduced Dosage Level 1 as per Table 1 . For patients previously on 1.9 mg/kg every 3 weeks, resume treatment at Reduced Dosage Level 2. If recurrent Grade 4 ocular toxicity is experienced, permanently discontinue BLENREP. Table 3. Recommended Dosage Modifications for Other Adverse Reactions a a Adverse reactions were graded according to the Common Terminology Criteria for Adverse Events v5.0. b Consider reverting to previous dose, if appropriate once platelet count recovers to 50,000/mcL or higher. Adverse Reaction Severity Recommended Dosage Modification Thrombocytopenia [see Warnings and Precautions ( 5.3 )] Platelet count between 25,000/mcL and 50,000/mcL without bleeding For patients on 2.5 mg/kg, reduce to Reduced Dosage Level 1 as per Table 1 . b For patients on 1.9 mg/kg, continue at same dosage. Platelet count between 25,000/mcL and 50,000/mcL with bleeding Withhold BLENREP until bleeding resolves. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1 . For patients on 1.9 mg/kg, resume at same dosage. Platelet count less than 25,000/mcL Withhold BLENREP until platelet count recovers to 25,000/mcL or higher. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1 . For patients on 1.9 mg/kg, resume at same dosage. Infusion-related Reactions Grade 2 Interrupt infusion and provide supportive care. Once symptoms resolve to Grade 1 or less, resume infusion at 50% of the initial rate prior to the event. Consider premedication for subsequent infusions. Grade 3 Interrupt infusion and provide supportive care. Once symptoms resolve to Grade 1 or less, resume at 50% of the initial rate prior to the event. Administer premedication for subsequent infusions. Grade 4 Permanently discontinue BLENREP. If anaphylactic or life‑threatening infusion reaction, permanently discontinue the infusion and institute appropriate emergency care. Other Adverse Reactions [see Adverse Reactions ( 6.1 ] Grade 3 Withhold BLENREP until adverse reaction improves to Grade 1 or less. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1 . For patients on 1.9 mg/kg, resume at same dosage. Grade 4 Consider permanent discontinuation of BLENREP. If continuing treatment, withhold BLENREP until adverse reaction improves to Grade 1 or less. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1 . For patients on 1.9 mg/kg, resume at same dosage.
How Supplied
BLENREP (belantamab mafodotin‑blmf) for injection is a sterile, preservative‑free, white to yellow lyophilized powder for reconstitution and further dilution prior to intravenous use. BLENREP is supplied in a carton containing one 70 mg single‑dose vial with a rubber stopper (not made with natural rubber latex) and aluminum overseal with removable cap (NDC 0173‑0913‑01). Store vials refrigerated at 36ºF to 46ºF (2ºC to 8ºC). BLENREP is a hazardous drug. Follow applicable special handling and disposal procedures. 1
Storage and Handling
BLENREP (belantamab mafodotin‑blmf) for injection is a sterile, preservative‑free, white to yellow lyophilized powder for reconstitution and further dilution prior to intravenous use. BLENREP is supplied in a carton containing one 70 mg single‑dose vial with a rubber stopper (not made with natural rubber latex) and aluminum overseal with removable cap (NDC 0173‑0913‑01). Store vials refrigerated at 36ºF to 46ºF (2ºC to 8ºC). BLENREP is a hazardous drug. Follow applicable special handling and disposal procedures. 1
Description
• BLENREP causes changes in the corneal epithelium resulting in changes in vision, including severe visual impairment, and symptoms such as blurred vision and dry eyes. In the clinical study, corneal ulcers, including cases with infection, also occurred [see Warnings and Precautions ( 5.1 )] . • Conduct ophthalmic exams at baseline, before each dose, promptly for new or worsening symptoms, and as clinically indicated. In the clinical study, 83% of patients required a dosage modification due to ocular toxicity. Withhold BLENREP until improvement and resume or permanently discontinue, based on severity [see Dosage and Administration ( 2.3 ), Warnings and Precautions ( 5.1 )] . • Because of the risk of ocular toxicity, BLENREP is available only through a restricted program called the BLENREP Risk Evaluation and Mitigation Strategy (REMS) [see Warnings and Precautions ( 5.2 )] .
Medication Information
Warnings and Precautions
• Thrombocytopenia: Monitor complete blood counts at baseline and periodically during treatment. Withhold or reduce the dosage based on severity. ( 2.3 , 5.3 ) • Embryo‑fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to fetus and to use effective contraception. ( 5.4 , 8.1 , 8.3 )
Indications and Usage
BLENREP is indicated in combination with bortezomib and dexamethasone for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least two prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent.
Dosage and Administration
• The recommended dosage of BLENREP, in combination with bortezomib and dexamethasone, is 2.5 mg/kg as an intravenous infusion over 30 minutes once every 3 weeks for 8 cycles, followed by BLENREP 2.5 mg/kg every 3 weeks as a single agent. ( 2.2 ) • See Full Prescribing Information for instructions on preparation and administration. ( 2.4 )
Contraindications
None.
Adverse Reactions
In the BVd arm of the clinical study, 98% of patients required a dosage modification for any component of treatment for an adverse reaction, including 87% who required a dosage modification of BLENREP [see Clinical Studies ( 14 )] . Eighty-three percent of patients required a dosage modification of BLENREP for ocular toxicity based on ophthalmic exam findings or other ocular adverse reactions as defined by the Common Terminology Criteria for Adverse Events (CTCAE) [see Adverse Reactions ( 6.1 )] . There were high rates of dosage modifications in early treatment cycles. By Cycle 3, 53% of patients had a dosage interruption or reduction, 7% had discontinued treatment, and only 40% received the planned dose of BLENREP. The recommended dosage modifications for ocular toxicity based on ophthalmic exam findings are provided in Tables 1 and 2. Ophthalmic exam findings include both corneal exam findings and change in BCVA as assessed by an eye care professional. The overall grade of ophthalmic exam findings is based on the worst finding in the worst affected eye, based on either corneal exam finding or a change in BCVA. Corneal exam findings may or may not be accompanied by changes in BCVA or ocular symptoms. • Do not re‑escalate the dose of BLENREP after a dosage reduction is made for ocular toxicity based on ophthalmic exam findings. • In the clinical study, 67% of patients required a dosage interruption of BLENREP for ocular toxicity that lasted longer than 3 weeks (time between doses, median: 5.7 weeks [range: 3 to 31 weeks]). The recommended dosage modifications for other adverse reactions, including dosage modifications for ocular adverse reactions based on the CTCAE, are provided in Table 3 . Table 1. Recommended Dosage Reductions of BLENREP for Adverse Reactions a Reduced Dosage Level 2 is specific to dosage reductions due to ocular toxicity based on ophthalmic exam findings. BLENREP Reduced Dosage Level 1 1.9 mg/kg every 3 weeks Reduced Dosage Level 2 a 1.9 mg/kg every 8 weeks Table 2. Recommended Dosage Modifications for Ocular Toxicity Based on Ophthalmic Exam Findings a BCVA = best‑corrected visual acuity. a Mild superficial keratopathy (documented worsening from baseline). Refer to Table 3 for recommended dosage modifications for other ocular adverse reactions. b Microcyst‑like deposits are considered at least a Grade 2 finding. Withhold BLENREP if any microcyst‑like deposits are observed. Severity Ophthalmic Exam Findings [see Warnings and Precautions ( 5.1 )] Recommended Dosage Modification Grade 1 Corneal Exam Findings: Mild superficial punctate keratopathy a and/or Change in BCVA: Decline from baseline of 1 line on Snellen Equivalent BCVA Continue treatment at current dosage. Grade 2 Corneal Exam Findings: Moderate superficial punctate keratopathy, patchy microcyst‑like deposits b , peripheral sub‑epithelial haze, or a new peripheral stromal opacity and/or Change in BCVA: Decline from baseline of 2 lines on Snellen Equivalent BCVA and not worse than 20/200 Withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less. Resume treatment at Reduced Dosage Level 1 as per Table 1 . If recurrent Grade 2 or 3 ocular toxicity is experienced, resume treatment at Reduced Dosage Level 2. Grade 3 Corneal Exam Findings: Severe superficial punctate keratopathy, diffuse microcyst‑like deposits b involving the central cornea, central sub‑epithelial haze, or a new central stromal opacity and/or Change in BCVA: Decline from baseline of 3 or more lines on Snellen Equivalent BCVA and not worse than 20/200 Grade 4 Corneal Exam Findings: Corneal epithelial defect or corneal ulcer, with or without infection and/or Change in BCVA: Decline to Snellen Equivalent BCVA of worse than 20/200 Consider permanent discontinuation of BLENREP. If continuing treatment, withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less. For patients previously on 2.5 mg/kg every 3 weeks, resume treatment at Reduced Dosage Level 1 as per Table 1 . For patients previously on 1.9 mg/kg every 3 weeks, resume treatment at Reduced Dosage Level 2. If recurrent Grade 4 ocular toxicity is experienced, permanently discontinue BLENREP. Table 3. Recommended Dosage Modifications for Other Adverse Reactions a a Adverse reactions were graded according to the Common Terminology Criteria for Adverse Events v5.0. b Consider reverting to previous dose, if appropriate once platelet count recovers to 50,000/mcL or higher. Adverse Reaction Severity Recommended Dosage Modification Thrombocytopenia [see Warnings and Precautions ( 5.3 )] Platelet count between 25,000/mcL and 50,000/mcL without bleeding For patients on 2.5 mg/kg, reduce to Reduced Dosage Level 1 as per Table 1 . b For patients on 1.9 mg/kg, continue at same dosage. Platelet count between 25,000/mcL and 50,000/mcL with bleeding Withhold BLENREP until bleeding resolves. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1 . For patients on 1.9 mg/kg, resume at same dosage. Platelet count less than 25,000/mcL Withhold BLENREP until platelet count recovers to 25,000/mcL or higher. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1 . For patients on 1.9 mg/kg, resume at same dosage. Infusion-related Reactions Grade 2 Interrupt infusion and provide supportive care. Once symptoms resolve to Grade 1 or less, resume infusion at 50% of the initial rate prior to the event. Consider premedication for subsequent infusions. Grade 3 Interrupt infusion and provide supportive care. Once symptoms resolve to Grade 1 or less, resume at 50% of the initial rate prior to the event. Administer premedication for subsequent infusions. Grade 4 Permanently discontinue BLENREP. If anaphylactic or life‑threatening infusion reaction, permanently discontinue the infusion and institute appropriate emergency care. Other Adverse Reactions [see Adverse Reactions ( 6.1 ] Grade 3 Withhold BLENREP until adverse reaction improves to Grade 1 or less. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1 . For patients on 1.9 mg/kg, resume at same dosage. Grade 4 Consider permanent discontinuation of BLENREP. If continuing treatment, withhold BLENREP until adverse reaction improves to Grade 1 or less. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1 . For patients on 1.9 mg/kg, resume at same dosage.
Storage and Handling
BLENREP (belantamab mafodotin‑blmf) for injection is a sterile, preservative‑free, white to yellow lyophilized powder for reconstitution and further dilution prior to intravenous use. BLENREP is supplied in a carton containing one 70 mg single‑dose vial with a rubber stopper (not made with natural rubber latex) and aluminum overseal with removable cap (NDC 0173‑0913‑01). Store vials refrigerated at 36ºF to 46ºF (2ºC to 8ºC). BLENREP is a hazardous drug. Follow applicable special handling and disposal procedures. 1
How Supplied
BLENREP (belantamab mafodotin‑blmf) for injection is a sterile, preservative‑free, white to yellow lyophilized powder for reconstitution and further dilution prior to intravenous use. BLENREP is supplied in a carton containing one 70 mg single‑dose vial with a rubber stopper (not made with natural rubber latex) and aluminum overseal with removable cap (NDC 0173‑0913‑01). Store vials refrigerated at 36ºF to 46ºF (2ºC to 8ºC). BLENREP is a hazardous drug. Follow applicable special handling and disposal procedures. 1
Description
• BLENREP causes changes in the corneal epithelium resulting in changes in vision, including severe visual impairment, and symptoms such as blurred vision and dry eyes. In the clinical study, corneal ulcers, including cases with infection, also occurred [see Warnings and Precautions ( 5.1 )] . • Conduct ophthalmic exams at baseline, before each dose, promptly for new or worsening symptoms, and as clinically indicated. In the clinical study, 83% of patients required a dosage modification due to ocular toxicity. Withhold BLENREP until improvement and resume or permanently discontinue, based on severity [see Dosage and Administration ( 2.3 ), Warnings and Precautions ( 5.1 )] . • Because of the risk of ocular toxicity, BLENREP is available only through a restricted program called the BLENREP Risk Evaluation and Mitigation Strategy (REMS) [see Warnings and Precautions ( 5.2 )] .
Section 42231-1
|
MEDICATION GUIDE BLENREP (BLEN-REP) (belantamab mafodotin-blmf) for injection, for intravenous use |
||
|
What is the most important information I should know about BLENREP? BLENREP can cause serious side effects, including:
|
||
|
|
|
|
Ulcers on the surface of the eye (corneal ulcers), including with infection, may also happen during treatment with BLENREP. Tell your healthcare provider right away if you notice any new or worsening eye symptoms or vision changes during treatment with BLENREP. Your healthcare provider will refer you to an eye care specialist (such as an ophthalmologist or optometrist) to check your eyes before you start treatment, before you receive each dose of BLENREP, and as needed for any new or worsening eye problems. It is important that you:
Because of the risk of eye problems, BLENREP is available only through a restricted program called the BLENREP Risk Evaluation and Mitigation Strategy (REMS). Your healthcare provider will give you the BLENREP REMS Patient Guide, which describes the risk of eye problems, and will explain the REMS program to you. To receive BLENREP, you must enroll in the REMS program and receive eye exams during treatment. See “What are the possible side effects of BLENREP?” for more information about side effects. |
||
|
What is BLENREP? BLENREP is a prescription medicine used in combination with the medicines bortezomib and dexamethasone to treat adults with multiple myeloma who:
It is not known if BLENREP is safe and effective in children. |
||
|
Before receiving BLENREP, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over‑the‑counter medicines, vitamins, and herbal supplements. |
||
|
How will I receive BLENREP?
|
||
|
What are the possible side effects of BLENREP? BLENREP can cause serious side effects, including:
The most common side effects of BLENREP when given with bortezomib and dexamethasone include: |
||
|
|
|
|
The most common severe abnormal blood test results during treatment with BLENREP include decreases in platelets, white blood cells, and hemoglobin, and increases in certain liver enzymes. Tell your healthcare provider right away if you get new or worsening unexplained signs or symptoms of lung problems, including shortness of breath, chest pain, or cough. Your healthcare provider may decrease your dose, temporarily stop, or completely stop treatment with BLENREP if you get serious side effects. BLENREP may affect fertility in males and females, which may affect your ability to have children. Talk to your healthcare provider if this is a concern for you. These are not all of the possible side effects of BLENREP. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‑800‑FDA‑1088. |
||
|
General information about the safe and effective use of BLENREP. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about BLENREP that is written for health professionals. |
||
|
What are the ingredients in BLENREP? Active Ingredient: belantamab mafodotin-blmf Inactive Ingredients: citric acid monohydrate, edetate disodium, polysorbate 80, sodium citrate, and trehalose. |
||
|
Manufactured by: GlaxoSmithKline LLC Philadelphia PA, 19104 U.S. License No. 1727 |
For: GlaxoSmithKline, Durham, NC 27701 ©2025 GSK group of companies or its licensor. BLP:1MG |
|
|
For more information, call GlaxoSmithKline (GSK) at 1‑888‑825‑5249. Trademarks are owned by or licensed to the GSK group of companies. |
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 10/2025
Section 51945-4
NDC 0173-0913-01
BLENREP
(belantamab mafodotin-blmf)
for injection
70 mg/vial
Rx Only
For intravenous infusion after reconstitution and dilution.
CAUTION: Hazardous Drug.
No preservative.
Dispense the enclosed Medication Guide to each patient.
Contains one Single-Dose vial.
Discard Unused Portion.
2025 GSK group of companies or its licensor.
Product Italy
Rev. 04/25
62000000097965
15 References
-
1.“OSHA Hazardous Drugs.” OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, BLENREP can cause fetal harm when administered to a pregnant woman, because it contains a genotoxic compound (the microtubule inhibitor, MMAF) and it targets actively dividing cells [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)]. Human immunoglobulin G (IgG) is known to cross the placenta; therefore, belantamab mafodotin-blmf has the potential to be transmitted from the mother to the developing fetus. There are no available data on the use of BLENREP in pregnant women to evaluate for drug-associated risk. No animal reproduction studies were conducted with BLENREP. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data: Animal reproductive or developmental toxicity studies were not conducted with belantamab mafodotin‑blmf. The cytotoxic component of BLENREP, mcMMAF, disrupts microtubule function, is genotoxic, and can be toxic to rapidly dividing cells, suggesting it has the potential to cause embryo‑fetal toxicity.
8.2 Lactation
Risk Summary
There are no data on the presence of belantamab mafodotin-blmf in human milk or the effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with BLENREP and for 3 months after the last dose.
11 Description
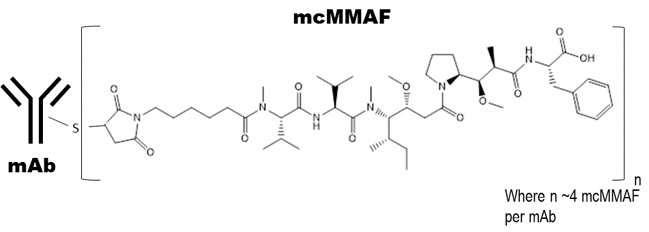
Belantamab mafodotin‑blmf is a B‑cell maturation antigen (BCMA)‑directed antibody and microtubule inhibitor conjugate. Belantamab mafodotin‑blmf is an antibody conjugate composed of 3 components: 1) afucosylated, humanized immunoglobulin G1 monoclonal antibody covalently linked to 2) the microtubule inhibitor mcMMAF via 3) a protease‑resistant maleimidocaproyl linker.
The antibody is produced in a mammalian cell line (Chinese Hamster Ovary) using recombinant DNA technology and the microtubule inhibitor and linker are produced by chemical synthesis. Approximately 4 molecules of mafodotin are attached to each antibody molecule. The molecular weight of belantamab mafodotin‑blmf is approximately 152 kDa. Belantamab mafodotin‑blmf has the following structure:
BLENREP (belantamab mafodotin‑blmf) for injection is a sterile, preservative‑free, white to yellow, lyophilized powder in a single‑dose vial for reconstitution and further dilution prior to intravenous use. BLENREP is supplied as 70 mg per vial and requires reconstitution with 1.4 mL of Sterile Water for Injection, USP, to obtain a concentration of 50 mg/mL. Each mL of reconstituted solution contains belantamab mafodotin‑blmf (50 mg) and the inactive ingredients, citric acid monohydrate (0.46 mg), edetate disodium (0.017 mg), polysorbate 80 (0.2 mg), sodium citrate (5.88 mg), and trehalose (68.4 mg). The pH of the reconstituted solution is 6.2.
8.4 Pediatric Use
The safety and effectiveness of BLENREP in pediatric patients have not been established.
8.5 Geriatric Use
Of 242 patients who received BLENREP in DREAMM-7, 121 patients (50%) were aged 65 years or older and 37 patients (15%) were 75 years or older. No overall differences in the safety of BLENREP were observed between patients 65 years of age and older and younger adult patients.
Of the 108 patients who received BLENREP and were evaluated for efficacy in the DREAMM-7 study, there was an insufficient number of older adult patients to determine if the effectiveness in patients 65 years of age and older is different than in younger adult patients.
12.6 Immunogenicity
The observed incidence of anti‑drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti‑drug antibodies in the studies described below with the incidence of anti‑drug antibodies (ADAs) in other studies, including those of BLENREP or of other belantamab mafodotin‑blmf products.
In the combination therapy studies, 3% of patients (15/515) tested positive for treatment‑emergent belantamab mafodotin‑blmf ADAs. Among the 15 patients who tested positive for ADAs, 2 patients tested positive for neutralizing anti‑belantamab mafodotin‑blmf antibodies.
14 Clinical Studies
Relapsed or Refractory Multiple Myeloma in Combination with Bortezomib and Dexamethasone
The efficacy of BLENREP in combination with bortezomib and dexamethasone (BVd) compared with daratumumab, bortezomib, and dexamethasone (DVd) was evaluated in DREAMM‑7 (NCT04246047), an open‑label, randomized, multicenter study in adult patients with relapsed or refractory multiple myeloma. Patients received at least one prior line of therapy and had documented disease progression during or after their most recent line of treatment. Patients who were refractory or intolerant to daratumumab or bortezomib, or with prior exposure to anti‑BCMA therapy were excluded. Patients with existing corneal disease, except for mild punctate keratopathy, were excluded.
Patients were randomized 1:1 to the following treatment arms:
-
•BLENREP 2.5 mg/kg (IV) every 3 weeks on Day 1 of each 21‑day cycle. Bortezomib 1.3 mg/m2 (subcutaneously) on Days 1, 4, 8, and 11 and dexamethasone 20 mg (IV or orally) on the day of, and the day after, bortezomib treatment of Cycles 1–8 (21‑day cycles). From Cycle 9 onward, treatment with BLENREP was continued as a single agent.
-
•Daratumumab 16 mg/kg (IV) every week for Cycles 1–3 and every 3 weeks for Cycles 4–8. Bortezomib 1.3 mg/m2 (subcutaneously) on Days 1, 4, 8, and 11 and dexamethasone 20 mg (IV or orally) on the day of, and the day after, bortezomib treatment of Cycles 1–8 (21‑day cycles). From Cycle 9 onward, treatment with daratumumab was continued as a single agent.
The dose level of dexamethasone in each arm was reduced by half in patients aged 75 years or older. Treatment with BLENREP or daratumumab was continued until disease progression or unacceptable toxicity.
Efficacy was established based on progression-free survival (PFS) and overall survival (OS).
A total of 217 patients who received at least two prior lines of therapy, including a proteasome inhibitor and immunomodulatory agent, were evaluated for efficacy: 108 in the BVd arm and 109 in the DVd arm. Baseline demographics and characteristics were similar across arms. The median age was 65 years (range: 39 to 86); 43% were age 65 to 74 years, 11% age 75 years or older; 53% were male; 86% were White, 11% Asian, 2% Black; R-ISS stage at screening was stage I in 37%, stage II in 56%, stage III in 6%; high‑risk cytogenetics (presence of t (11;14), t (14;16) or 17p13del) were present in 29%; extramedullary disease was present in 11%. The median number of prior lines of therapy was 3 (range: 2 to 7); 2% received a prior anti-CD38 monoclonal antibody, and 71% previously received autologous stem cell transplantation. Overall, 19% of patients were refractory to proteasome inhibitors and 59% were refractory to immunomodulatory agents.
Efficacy results are summarized in Table 8 and Figures 1 and 2.
|
BLENREP +
Bortezomib and Dexamethasone N = 108 |
Daratumumab +
Bortezomib and Dexamethasone N = 109 |
|
|---|---|---|
| NR = not reached. a Based on all randomized patients who received at least two prior lines of therapy, including a proteasome inhibitor and immunomodulatory agent. b Median follow-up of 27.9 months. c Response was based on Independent Review Committee per International Myeloma Working Group criteria. d By Brookmeyer and Crowley method. e Based on stratified Cox regression model. f Median follow-up of 38.7 months. g ORR: sCR+CR+VGPR+PR. h Assessed by next generation sequencing assay (clonoSEQ) at 10‑5 threshold. |
||
|
Progression‑Free Survival (PFS)a,b,c |
||
|
Number (%) of patients with event |
42 (39) |
73 (67) |
|
Median in months (95% CI)d |
31.3 (23.5, NR) |
10.4 (7, 13.4) |
|
Hazard ratio (95% CI)e |
0.31 (0.21, 0.47) |
|
|
Overall Survival (OS)a,f |
||
|
Number (%) of patients with event |
33 (31) |
57 (52) |
|
Median in months (95% CI)d |
NR (NR, NR) |
35.7 (21.1, NR) |
|
Hazard ratio (95% CI)e |
0.49 (0.32, 0.76) |
|
|
Overall Response Rate (ORR)a,b,c,g, % (95% CI) |
81.5 (72.9, 88.3) |
56.9 (47, 66.3) |
|
Stringent complete response (sCR), n (%) |
15 (13.9) |
4 (3.7) |
|
Complete response (CR), n (%) |
19 (17.6) |
5 (4.6) |
|
Very good partial response (VGPR), n (%) |
34 (31.5) |
23 (21.1) |
|
Partial response (PR), n (%) |
20 (18.5) |
30 (27.5) |
|
Minimal Residual Disease (MRD) Negativity Ratea,b,h, n (%) |
25 (23.1) |
3 (2.8) |
|
95% CI |
(15.6, 32.2) |
(0.6, 7.8) |
|
MRD Negativity Rate in Patients with CR or Betterb,h |
||
|
Number of patients with CR or better |
34 |
9 |
|
MRD negativity rate, n (%) |
25 (73.5) |
3 (33.3) |
|
(95% CI) |
(55.6, 87.1) |
(7.5, 70.1) |
Figure 1: Kaplan-Meier Curve for Progression‑Free Survival in DREAMM-7
Figure 2: Kaplan-Meier Curve for Overall Survival in DREAMM-7
In patients who achieved response, the median time to response was 1.43 months (range: 0.7 to 8.4 months) in the BVd arm and 1.03 months (range: 0.7 to 11.1 months) in the DVd arm.
4 Contraindications
None.
5.1 Ocular Toxicity
BLENREP causes ocular toxicity, defined as changes in the corneal epithelium and changes in BCVA based on ophthalmic exam (including slit lamp exam), or other ocular adverse reactions as defined by the CTCAE [see Adverse Reactions (6.1)].
In DREAMM-7, ocular toxicity occurred in 92% of patients, including Grade 3 or 4 in 77% of patients. The most common ocular toxicities (>25%) were reduction in BCVA (89%) and corneal exam findings (86%) based on ophthalmic exam findings, blurred vision (66%), dry eye (51%), photophobia (47%), foreign body sensation in eyes (44%), eye irritation (43%), and eye pain (33%) [see Adverse Reactions (6.1)].
Ocular toxicity based on ophthalmic exam findings was reported as Grade 2 in 9% of patients, Grade 3 in 56% of patients, and Grade 4 in 21% of patients. The median time to onset of the first Grade 2 to 4 ophthalmic exam findings was 43 days (range: 15 to 611 days). The median duration of all Grade 2 to 4 ophthalmic exam findings was 85 days (range: 5 to 813 days). Patients experienced a median of 3 episodes (range: 1 to 11 episodes) of ocular toxicity based on ophthalmic exam findings. Of the patients with Grade 2 to 4 ophthalmic exam findings, 42% had improvement of the last event to Grade 1 or better; 22% had resolution of the last event based on return to baseline or normal ophthalmic exam findings.
The most commonly reported corneal exam findings included superficial punctate keratopathy, microcyst-like deposits, epithelial changes, and haze. Cases of corneal ulcer, including cases with infection, have been reported and should be managed promptly by an eye care professional [see Adverse Reactions (6.1)].
A reduction in BCVA to 20/50 or worse in at least one eye occurred in 69% of patients, including 29% who experienced a change in BCVA to 20/100 or worse, and 12% who experienced a change in BCVA to 20/200 or worse. Of the patients with reduced BCVA to 20/50 or worse in at least one eye, 61% had resolution of the last event to baseline or better. Of the patients with reduced BCVA to 20/100 or worse, 57% had resolution of the last event. Of the patients with reduced BCVA to 20/200 or worse, 48% had resolution of the last event.
Ophthalmic exams (including slit lamp exam and BCVA assessment) should be conducted by an eye care professional, such as an ophthalmologist or optometrist, at baseline, before each dose of BLENREP, promptly for new or worsening symptoms, and as clinically indicated. Perform baseline exam within 4 weeks prior to the first dose. Perform each follow-up exam within 10 days prior to the next planned dose. All effort should be made to schedule the exam as close to BLENREP dosing as possible. Withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less and resume at same or reduced dose or permanently discontinue based on severity [see Dosage and Administration (2.1, 2.3)].
Counsel patients to promptly inform their healthcare provider of any ocular symptoms. Counsel patients to use preservative‑free artificial tears at least 4 times a day starting with the first infusion and continuing until the end of treatment, and to avoid wearing contact lenses for the duration of therapy. Bandage contact lenses may be used under the direction of an eye care professional [see Dosage and Administration (2.1)].
Changes in visual acuity may be associated with difficulty for driving and reading. Counsel patients to use caution when driving or operating machinery.
BLENREP is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) [see Warnings and Precautions (5.2)].
6 Adverse Reactions
5.3 Thrombocytopenia
Thrombocytopenia of any grade occurred in 100% of patients in DREAMM‑7 [see Adverse Reactions (6.1)].
Grade 2 thrombocytopenia occurred in 10% of patients, Grade 3 in 29% of patients, and Grade 4 in 45% of patients. Clinically significant bleeding (Grade ≥2) occurred in 7% of patients with concomitant low platelet levels (Grade 3 or 4).
Monitor complete blood cell counts at baseline and periodically during treatment as clinically indicated. Withhold or reduce the dose of BLENREP based on severity [see Dosage and Administration (2.3)].
12.2 Pharmacodynamics
Exposure‑Response Relationships
When BLENREP was used in combination with Vd, higher belantamab mafodotin‑blmf Cycle 1 exposure was associated with a higher incidence of some safety adverse reactions (e.g., Grade ≥2 corneal exam findings).
Cardiac Electrophysiology
Belantamab mafodotin‑blmf had no meaningful QTc prolongation (>10 ms) at the recommended dosage.
12.3 Pharmacokinetics
Belantamab mafodotin‑blmf exhibited dose‑proportional pharmacokinetics, with a gradual decrease in clearance over time. After a planned infusion duration of 30 minutes, maximum belantamab mafodotin‑blmf plasma concentrations occurred at or shortly after the end of the infusion. Accumulation of belantamab mafodotin‑blmf was minimal to moderate for the ADC (Cycle 3 to Cycle 1 ratio was 1.13 for Cmax and 1.58 for AUC) as observed with a dosing regimen of every 3 weeks.
Table 7 describes the pharmacokinetics of belantamab mafodotin‑blmf for the 2.5 mg/kg dose on Cycle 1 at the end of the first 3‑week intervals.
| AUCb | Cmax | Ctau | |
|---|---|---|---|
| ADC = antibody drug conjugate; AUC = area under the curve; Cmax = maximum plasma concentration; Ctau = concentration at the end of a dosing interval. a Data presented as geometric mean (CV%). b AUC for ADC is AUC(0‑21days) and for cys-mcMMAF is AUC(0‑7days). |
|||
|
ADC (%) |
3,950 mcg•h/mL (30.6) |
43.7 mcg/mL (22.1) |
2.03 mcg/mL (62.5) |
|
cys‑mcMMAF (%) |
94.2 ng•h/mL (42.3) |
0.976 ng/mL (45.3) |
– |
Distribution
In vitro, cys‑mcMMAF exhibited low protein binding (70% unbound at a concentration of 5 ng/mL) in human plasma.
The geometric mean (CV%) steady‑state volume of distribution of belantamab mafodotin‑blmf was 10.8 L (22.2%).
Elimination
The geometric mean (CV%) belantamab mafodotin‑blmf (ADC) initial systemic clearance (CL) was 0.901 L/day (40%), and the elimination half‑life was 13 days (26%). Following treatment, steady‑state CL was 0.605 L/day (43%) or approximately 33% lower than initial systemic CL with an elimination half‑life of 17 days (31%).
The fraction of intact cys‑mcMMAF excreted in urine was approximately 18% of the dose in Cycle 1, with no evidence of other mcMMAF‑related metabolites.
Metabolism: The monoclonal antibody portion of belantamab mafodotin‑blmf is expected to undergo proteolysis to small peptides and individual amino acids by ubiquitous proteolytic enzymes. Cys‑mcMMAF had limited metabolic clearance in human hepatic S9 fraction incubation studies.
Specific Populations
No clinically significant differences in the pharmacokinetics of belantamab mafodotin‑blmf were observed based on age (32 to 89 years), sex, race (White vs. Black vs. Asian), body weight (37 to 170 kg), mild to severe renal impairment and kidney failure (eGFR <30 mL/min), or mild hepatic impairment (total bilirubin >ULN to ≤1.5 × ULN and any AST or total bilirubin ≤ULN with AST >ULN).
The effects of moderate (total bilirubin >1.5 × ULN to ≤3 × ULN and any AST) or severe hepatic impairment (total bilirubin >3 × ULN and any AST) on the pharmacokinetics of belantamab mafodotin‑blmf are unknown.
In Vitro Studies:
Cytochrome P450 (CYP) Enzymes: Cys-mcMMAF is not an inhibitor, an inducer, or a sensitive substrate of cytochrome P450 enzymes.
Transporter Systems: Cys‑mcMMAF is a substrate of organic anion transporting polypeptide (OATP)1B1 and OATP1B3, multidrug resistance‑associated protein (MRP)1, MRP2, and MRP3, bile salt export pump (BSEP), and a possible substrate of P‑glycoprotein (P‑gp).
2.2 Recommended Dosage
The recommended dosage for BLENREP is 2.5 mg/kg of actual body weight once every 3 weeks in combination with bortezomib and dexamethasone (BVd) for the first 8 cycles, followed by BLENREP 2.5 mg/kg of actual body weight once every 3 weeks as a single agent until disease progression or unacceptable toxicity.
For dosing instructions of agents administered in combination with BLENREP, see Clinical Studies (14) and respective Prescribing Information, as appropriate.
BLENREP is administered as an intravenous infusion over approximately 30 minutes.
8.6 Hepatic Impairment
No dose adjustment is recommended for patients with mild hepatic impairment (total bilirubin > upper limit of normal [ULN] to ≤1.5 × ULN and any aspartate aminotransferase [AST] or total bilirubin ≤ULN with AST >ULN).
The recommended dose of BLENREP has not been established in patients with moderate or severe hepatic impairment [see Clinical Pharmacology (12.3)].
1 Indications and Usage
BLENREP is indicated in combination with bortezomib and dexamethasone for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least two prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent.
12.1 Mechanism of Action
Belantamab mafodotin‑blmf is an antibody‑drug conjugate (ADC). The antibody component is an afucosylated IgG1 directed against BCMA, a protein expressed on normal B lymphocytes and multiple myeloma cells. The small molecule component is mcMMAF, a microtubule inhibitor. Upon binding to BCMA, belantamab mafodotin‑blmf is internalized followed by release of the cytotoxic agent (cys‑mcMMAF) via proteolytic cleavage. The released cys‑mcMMAF intracellularly disrupts the microtubule network, leading to cell cycle arrest and apoptosis.
Belantamab mafodotin‑blmf had antitumor activity in multiple myeloma cells and mediated killing of tumor cells through cys‑mcMMAF‑induced apoptosis, as well as by tumor cell lysis through antibody‑dependent cellular cytotoxicity and antibody‑dependent cellular phagocytosis.
Warning: Ocular Toxicity
-
•BLENREP causes changes in the corneal epithelium resulting in changes in vision, including severe visual impairment, and symptoms such as blurred vision and dry eyes. In the clinical study, corneal ulcers, including cases with infection, also occurred [see Warnings and Precautions (5.1)].
-
•Conduct ophthalmic exams at baseline, before each dose, promptly for new or worsening symptoms, and as clinically indicated. In the clinical study, 83% of patients required a dosage modification due to ocular toxicity. Withhold BLENREP until improvement and resume or permanently discontinue, based on severity [see Dosage and Administration (2.3), Warnings and Precautions (5.1)].
-
•Because of the risk of ocular toxicity, BLENREP is available only through a restricted program called the BLENREP Risk Evaluation and Mitigation Strategy (REMS) [see Warnings and Precautions (5.2)].
5.4 Embryo Fetal Toxicity
Based on its mechanism of action, BLENREP can cause fetal harm when administered to a pregnant woman because it contains a genotoxic compound (the microtubule inhibitor, monomethyl auristatin F [MMAF]) and it targets actively dividing cells.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with BLENREP and for 4 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with BLENREP and for 6 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
5 Warnings and Precautions
-
•Thrombocytopenia: Monitor complete blood counts at baseline and periodically during treatment. Withhold or reduce the dosage based on severity. (2.3, 5.3)
-
•Embryo‑fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to fetus and to use effective contraception. (5.4, 8.1, 8.3)
2 Dosage and Administration
-
•The recommended dosage of BLENREP, in combination with bortezomib and dexamethasone, is 2.5 mg/kg as an intravenous infusion over 30 minutes once every 3 weeks for 8 cycles, followed by BLENREP 2.5 mg/kg every 3 weeks as a single agent. (2.2)
-
•See Full Prescribing Information for instructions on preparation and administration. (2.4)
3 Dosage Forms and Strengths
For injection: 70 mg of belantamab mafodotin-blmf as a white to yellow lyophilized powder in a single-dose vial for reconstitution and further dilution.
8 Use in Specific Populations
Lactation: Advise not to breastfeed. (8.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Relapsed or Refractory Multiple Myeloma in Combination with Bortezomib and Dexamethasone
The safety of BLENREP with bortezomib and dexamethasone (n = 242) compared with daratumumab with bortezomib and dexamethasone (n = 246) was evaluated in DREAMM‑7 in patients with relapsed or refractory multiple myeloma who received at least one prior line of therapy [see Clinical Studies (14)]. Patients received BLENREP 2.5 mg/kg of actual body weight once every 3 weeks in combination with bortezomib and dexamethasone (BVd) for the first 8 cycles, followed by BLENREP as a single agent or daratumumab in combination with bortezomib and dexamethasone (DVd) for the first 8 cycles, followed by daratumumab as a single agent. Among patients who received BLENREP, 69% were exposed for 6 months or longer and 55% were exposed for greater than one year. The safety of BLENREP in combination with bortezomib and dexamethasone in patients who received only one prior line of therapy (n = 125) has not been established.
Serious adverse reactions occurred in 50% of patients who received BVd. Serious adverse reactions in ≥2% of patients included pneumonia (18%), pyrexia (5%), thrombocytopenia (5%), COVID-19 (5%), upper respiratory tract infection (4%), sepsis (4%), second primary malignancy (3%), and anemia (2%). Fatal adverse reactions occurred in 10% of patients who received BVd. Fatal adverse reactions which occurred in >1 patient included pneumonia (4%), sepsis (2%), COVID-19 (1%), respiratory failure (<1%), and intracranial hemorrhage (<1%).
Permanent discontinuation of BLENREP due to an adverse reaction occurred in 17% of patients. Adverse reactions which resulted in permanent discontinuation of BLENREP in ≥3% of patients included ocular toxicity (9%) and pneumonia (4%).
Dosage interruptions of BLENREP due to an adverse reaction occurred in 78% of patients. Adverse reactions which required dosage interruption of BLENREP in ≥3% of patients included ocular toxicity based on ophthalmic exam findings (74%), blurred vision (32%), upper respiratory tract infection (20%), dry eye (14%), photophobia (14%), pneumonia (14%), eye irritation (13%), COVID-19 (12%), foreign body sensation in eyes (12%), eye pain (10%), thrombocytopenia (9%), visual impairment (7%), cataract (5%), diarrhea (4%), and neutropenia (4%).
Dosage reductions of BLENREP due to an adverse reaction occurred in 36% of patients. Adverse reactions which required dosage reductions for BLENREP in ≥3% of patients included ocular toxicity based on ophthalmic exam findings (30%), thrombocytopenia (14%), and blurred vision (10%).
The most common adverse reactions (≥20%) were reduction in BCVA, corneal exam findings, blurred vision, dry eye, photophobia, foreign body sensation in eyes, eye irritation, upper respiratory tract infection, hepatotoxicity, eye pain, diarrhea, fatigue, pneumonia, cataract, and COVID-19. The most common Grade 3 or 4 laboratory abnormalities (≥10%) were decreased platelets, decreased lymphocytes, decreased neutrophils, increased gamma glutamyltransferase, decreased white blood cells, and decreased hemoglobin.
Table 4 summarizes the adverse reactions in DREAMM‑7.
| BCVA = best‑corrected visual acuity; BVd = BLENREP + bortezomib and dexamethasone; DVd = daratumumab + bortezomib and dexamethasone. a Adverse reactions, except ophthalmic exam findings, were graded according to Common Terminology Criteria for Adverse Events v5.0. b Based on ophthalmic exam findings. c Grouped term includes other related terms. d Includes the following fatal adverse reactions: BVd: pneumonia (n = 10), COVID-19 (n = 3), upper respiratory tract infection (n = 1); DVd: pneumonia (n = 7), COVID-19 (n = 5), pyrexia (n = 1). |
||||
|
Adverse Reactiona |
BLENREP + Bortezomib and Dexamethasone N = 242 |
Daratumumab + Bortezomib and Dexamethasone N = 246 |
||
|
All Grades (%) |
Grades 3‑4 (%) |
All Grades (%) |
Grades 3‑4 (%) |
|
|
Eye disorders |
||||
|
Reduction in BCVAb |
89 |
57 |
44 |
9 |
|
Corneal exam findingsb |
86 |
72 |
19 |
3 |
|
Blurred vision |
66 |
22 |
11 |
0.8 |
|
Dry eyec |
51 |
7 |
7 |
0 |
|
Photophobia |
47 |
2 |
2 |
0 |
|
Foreign body sensation in eyesc |
44 |
3 |
4 |
0 |
|
Eye irritation |
43 |
5 |
5 |
0 |
|
Eye painc |
33 |
0.8 |
4 |
0.4 |
|
Cataractc |
24 |
8 |
14 |
3 |
|
Visual impairment |
11 |
5 |
2 |
0.4 |
|
Gastrointestinal disorders |
||||
|
Diarrhea |
32 |
4 |
31 |
4 |
|
Nausea |
16 |
0.8 |
12 |
0 |
|
Infections |
||||
|
Upper respiratory tract infectionc,d |
38 |
2 |
36 |
2 |
|
Pneumoniac,d |
26 |
16 |
17 |
5 |
|
COVID-19d |
24 |
5 |
20 |
2 |
|
Hepatobiliary disorders |
||||
|
Hepatotoxicityc |
33 |
14 |
16 |
2 |
|
General disorders and administration site conditions |
||||
|
Fatiguec |
26 |
6 |
27 |
4 |
|
Pyrexiac,d |
19 |
0.4 |
11 |
2 |
Clinically relevant adverse reactions in <10% of patients who received BVd included: increased lacrimation, vomiting, diplopia, albuminuria, sepsis, eye pruritus, infusion-related reactions, corneal ulcer (including cases with infection), and pneumonitis.
Table 5 summarizes the laboratory abnormalities in DREAMM-7.
| Laboratory Abnormality |
BLENREP +
Bortezomib and Dexamethasonea |
Daratumumab +
Bortezomib and Dexamethasonea |
||
|---|---|---|---|---|
|
All Grades
(%) |
Grades 3‑4
(%) |
All Grades
(%) |
Grades 3‑4
(%) |
|
| BVd = BLENREP + bortezomib and dexamethasone; DVd = daratumumab + bortezomib and dexamethasone. a The denominator used to calculate the rate varied from 238 to 241 (BVd) and 243 to 246 (DVd) based on the number of patients with a baseline value and at least one post-treatment value. |
||||
|
Hematology |
||||
|
Platelets decreased |
100 |
74 |
88 |
48 |
|
Lymphocytes decreased |
90 |
53 |
92 |
56 |
|
Leukocytes decreased |
59 |
11 |
67 |
17 |
|
Neutrophils decreased |
52 |
17 |
53 |
13 |
|
Hemoglobin decreased |
51 |
10 |
60 |
12 |
|
Chemistry |
||||
|
Aspartate aminotransferase increased |
88 |
5 |
40 |
0 |
|
Gamma glutamyltransferase increased |
73 |
15 |
44 |
3 |
|
Alanine aminotransferase increased |
71 |
5 |
54 |
1 |
|
Creatinine increased |
51 |
2 |
53 |
<1 |
|
Creatine phosphokinase increased |
48 |
3 |
31 |
3 |
Patient-reported ocular symptoms were assessed using the Patient-Reported Outcomes Common Terminology Criteria for Adverse Events (PRO-CTCAE) and Ocular Surface Disease Index (OSDI).
PRO-CTCAE assessments were collected at baseline and then every 3 weeks until treatment discontinuation. Completion rates in both arms were ≥90% at baseline and ≥81% at subsequent timepoints where >50% of patients remained on treatment. OSDI assessments were collected every 3 weeks until the 6th dose and then every 6 weeks thereafter until treatment discontinuation for BVd and every 3 weeks until Cycle 6 and then every 12 weeks thereafter until treatment discontinuation for DVd. Completion rates in both arms were ≥90% at baseline and ≥52% at subsequent timepoints where >50% of patients remained on treatment.
Table 6 summarizes the patient-reported symptom of blurred vision as assessed by PRO-CTCAE.
| Symptom (Attribute)a | Any Symptom Before Treatmentb | Score 3 or 4 Before Treatmentc | Any Symptom on Treatmentd,e | Score 3 or 4 on Treatmentd,f | ||||
|---|---|---|---|---|---|---|---|---|
|
BVd (%)
n = 232 |
DVd (%)
n = 227 |
BVd (%)
n = 232 |
DVd (%)
n = 227 |
BVd (%)
n = 238 |
DVd (%)
n = 239 |
BVd (%)
n = 238 |
DVd (%)
n = 239 |
|
| BVd = BLENREP + bortezomib and dexamethasone; DVd = daratumumab + bortezomib and dexamethasone; PRO-CTCAE = patient-reported outcomes common terminology criteria for adverse events. a Symptom attribute scoring defined as severity with a score of 0 = ‘none’; 1 = ‘mild’; 2 = ‘moderate’; 3 = ‘severe’; 4 = ‘very severe’. b Percentage of patients whose symptom score before treatment was 1 to 4. c Percentage of patients whose symptom score before treatment was 3 or 4. d Number of patients who provided at least one on-treatment score. e Percentage of patients whose maximum post-baseline score was 1 to 4 on treatment. f Percentage of patients whose maximum post-baseline score was 3 or 4 on treatment. |
||||||||
|
Blurred vision (severity) |
28 |
24 |
<1 |
2 |
93 |
74 |
55 |
15 |
Driving at night was assessed using the OSDI. At baseline, the proportion of patients who reported limitations with driving at night “all of the time” or “most of the time” during the last week was 9% in the BVd arm and 4% in the DVd arm. The proportion of patients who reported limitations with driving at night “all of the time” or “most of the time” during the last week was highest in the BVd arm at 47% at Week 13 and in the DVd arm at 12% at Week 76.
2.1 Important Safety Information
Ophthalmic exams, including slit lamp exam and assessment of best‑corrected visual acuity (BCVA), should be conducted by an eye care professional, such as an ophthalmologist or optometrist. Conduct ophthalmic exams at baseline, before each dose of BLENREP, promptly for new or worsening symptoms, and as clinically indicated [see Warnings and Precautions (5.1)].
Counsel patients to promptly inform their healthcare provider of any ocular symptoms. Advise patients to use preservative‑free artificial tears at least 4 times a day starting with the first infusion and continuing until end of treatment, and to avoid wearing contact lenses for the duration of therapy. Bandage contact lenses may be used under the direction of an eye care professional [see Warnings and Precautions (5.1)].
17 Patient Counseling Information
Advise the patient to read the FDA‑approved patient labeling (Medication Guide).
Ocular Toxicity
-
•Advise patients that ocular toxicity may occur during treatment with BLENREP [see Warnings and Precautions (5.1)].
-
•Advise patients to promptly tell their healthcare provider if they notice any new or worsening eye symptoms [see Dosage and Administration (2.1), Warnings and Precautions (5.1)].
-
•Advise patients that they will be sent to an eye care professional to obtain ophthalmic exams before starting BLENREP, before each dose, promptly for any new or worsening eye symptoms, and as clinically indicated [see Dosage and Administration (2.1), Warnings and Precautions (5.1)].
-
•Advise patients to administer preservative‑free artificial tears at least 4 times per day starting with the first infusion and continuing until the end of treatment and to avoid wearing contact lenses for the duration of therapy. Bandage contact lenses may be used under the direction of an eye care professional [see Dosage and Administration (2.1, 2.3), Warnings and Precautions (5.1)].
-
•Advise patients to use caution when driving or operating machinery as BLENREP may adversely affect their vision [see Warnings and Precautions (5.1)].
BLENREP REMS
-
•Advise patients that because of the risk of ocular toxicity, BLENREP is available only through a restricted program called the BLENREP REMS [see Warnings and Precautions (5.2)].
-
•Patients must receive counseling about the risk of ocular toxicity, enroll in the REMS, and adhere to ongoing monitoring via ophthalmic exams. Patients will be given the BLENREP REMS Patient Guide. This guide describes the risk of ocular toxicity with BLENREP [see Warnings and Precautions (5.2)].
Thrombocytopenia
-
•Advise patients to inform their healthcare provider if they develop signs or symptoms of bleeding [see Warnings and Precautions (5.3)].
Embryo‑fetal Toxicity
-
•Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4), Use in Specific Populations (8.1, 8.3)].
-
•Advise females of reproductive potential to use effective contraception during treatment with BLENREP and for 4 months after the last dose [see Warnings and Precautions (5.4), Use in Specific Populations (8.3)].
-
•Advise males with female partners of reproductive potential to use effective contraception during treatment with BLENREP and for 6 months after the last dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
-
•Advise women not to breastfeed during treatment with BLENREP and for 3 months after the last dose [see Use in Specific Populations (8.2)].
Infertility
-
•Advise males and females of reproductive potential that BLENREP may impair fertility [see Use in Specific Populations (8.3)].
Pneumonitis
-
•Advise patients to immediately report any new or worsening respiratory symptoms to their healthcare provider [see Adverse Reactions (6.1)].
Trademarks are owned by or licensed to the GSK group of companies.
Manufactured by:
GlaxoSmithKline LLC
Philadelphia PA, 19104
U.S. License No. 1727
For:
GlaxoSmithKline
Durham, NC 27701
©2025 GSK group of companies or its licensor.
BLP:1PI
2.4 Preparation and Administration
BLENREP is a hazardous drug. Follow applicable special handling and disposal procedures.1
Calculate the dose (mg), total volume (mL) of solution required, and the number of vials of BLENREP needed based on the patient’s actual body weight. More than one vial may be needed for a full dose.
Reconstitution
-
•Remove the vial(s) of BLENREP from the refrigerator and allow to stand for approximately 10 minutes to reach room temperature (68°F to 77°F [20°C to 25°C]).
-
•Reconstitute each 70 mg vial of BLENREP with 1.4 mL of Sterile Water for Injection, USP, to obtain a final concentration of 50 mg/mL. Gently swirl the vial to aid dissolution. Do not shake.
-
•If the reconstituted solution is not used immediately, store in the original container refrigerated at 36ºF to 46ºF (2ºC to 8ºC) or at room temperature (68°F to 77°F [20°C to 25°C]) for up to 4 hours. Discard if not diluted within 4 hours. Do not freeze.
-
•Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The reconstituted solution should be a clear to opalescent, colorless to yellow to brown liquid. Discard if extraneous particulate matter is observed.
Dilution
-
•Withdraw the calculated volume of BLENREP from the appropriate number of vials and dilute in a 250 mL infusion bag of 0.9% Sodium Chloride Injection, USP, to a final concentration of 0.2 mg/mL to 2 mg/mL. The infusion bag must be made of polyvinylchloride (PVC) or polyolefin (PO).
-
•Mix the diluted solution by gentle inversion. Do not shake.
-
•Discard any unused reconstituted solution of BLENREP left in the vial(s).
-
•If the diluted infusion solution is not used immediately, store refrigerated at 36ºF to 46ºF (2ºC to 8ºC) for up to 24 hours. Do not freeze. Once removed from refrigeration, administer the diluted infusion solution of BLENREP within 6 hours (including infusion time).
-
•Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The diluted infusion solution should be clear and colorless. Discard if particulate matter is observed.
Administration
-
•If refrigerated, allow the diluted infusion solution to equilibrate to room temperature (68ºF to 77ºF [20ºC to 25ºC]) prior to administration. Diluted infusion solution may be kept at room temperature for no more than 6 hours (including infusion time).
-
•Administer by intravenous infusion over approximately 30 minutes using an infusion set made of PVC or PO.
-
•Filtration of the diluted solution is not required; however, if the diluted solution is filtered, use a polyethersulfone (PES)-based filter (0.2 micron).
Do not mix or administer BLENREP with other products. The product does not contain a preservative.
16 How Supplied/storage and Handling
BLENREP (belantamab mafodotin‑blmf) for injection is a sterile, preservative‑free, white to yellow lyophilized powder for reconstitution and further dilution prior to intravenous use.
BLENREP is supplied in a carton containing one 70 mg single‑dose vial with a rubber stopper (not made with natural rubber latex) and aluminum overseal with removable cap (NDC 0173‑0913‑01).
Store vials refrigerated at 36ºF to 46ºF (2ºC to 8ºC).
BLENREP is a hazardous drug. Follow applicable special handling and disposal procedures.1
2.3 Dosage Modifications for Adverse Reactions
In the BVd arm of the clinical study, 98% of patients required a dosage modification for any component of treatment for an adverse reaction, including 87% who required a dosage modification of BLENREP [see Clinical Studies (14)]. Eighty-three percent of patients required a dosage modification of BLENREP for ocular toxicity based on ophthalmic exam findings or other ocular adverse reactions as defined by the Common Terminology Criteria for Adverse Events (CTCAE) [see Adverse Reactions (6.1)]. There were high rates of dosage modifications in early treatment cycles. By Cycle 3, 53% of patients had a dosage interruption or reduction, 7% had discontinued treatment, and only 40% received the planned dose of BLENREP.
The recommended dosage modifications for ocular toxicity based on ophthalmic exam findings are provided in Tables 1 and 2. Ophthalmic exam findings include both corneal exam findings and change in BCVA as assessed by an eye care professional. The overall grade of ophthalmic exam findings is based on the worst finding in the worst affected eye, based on either corneal exam finding or a change in BCVA. Corneal exam findings may or may not be accompanied by changes in BCVA or ocular symptoms.
-
•Do not re‑escalate the dose of BLENREP after a dosage reduction is made for ocular toxicity based on ophthalmic exam findings.
-
•In the clinical study, 67% of patients required a dosage interruption of BLENREP for ocular toxicity that lasted longer than 3 weeks (time between doses, median: 5.7 weeks [range: 3 to 31 weeks]).
The recommended dosage modifications for other adverse reactions, including dosage modifications for ocular adverse reactions based on the CTCAE, are provided in Table 3.
| a Reduced Dosage Level 2 is specific to dosage reductions due to ocular toxicity based on ophthalmic exam findings. | |
|
BLENREP |
|
|
Reduced Dosage Level 1 |
1.9 mg/kg every 3 weeks |
|
Reduced Dosage Level 2a |
1.9 mg/kg every 8 weeks |
| BCVA = best‑corrected visual acuity. a Mild superficial keratopathy (documented worsening from baseline). Refer to Table 3 for recommended dosage modifications for other ocular adverse reactions. b Microcyst‑like deposits are considered at least a Grade 2 finding. Withhold BLENREP if any microcyst‑like deposits are observed. |
||
|
Severity |
Ophthalmic Exam Findings [see Warnings and Precautions (5.1)] |
Recommended Dosage Modification |
|
Grade 1 |
Corneal Exam Findings:
and/or Change in BCVA:
|
Continue treatment at current dosage. |
|
Grade 2 |
Corneal Exam Findings:
and/or Change in BCVA:
|
Withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less. Resume treatment at Reduced Dosage Level 1 as per Table 1. If recurrent Grade 2 or 3 ocular toxicity is experienced, resume treatment at Reduced Dosage Level 2. |
|
Grade 3 |
Corneal Exam Findings:
and/or Change in BCVA:
|
|
|
Grade 4 |
Corneal Exam Findings:
and/or Change in BCVA:
|
Consider permanent discontinuation of BLENREP. If continuing treatment, withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less. For patients previously on 2.5 mg/kg every 3 weeks, resume treatment at Reduced Dosage Level 1 as per Table 1. For patients previously on 1.9 mg/kg every 3 weeks, resume treatment at Reduced Dosage Level 2. If recurrent Grade 4 ocular toxicity is experienced, permanently discontinue BLENREP. |
|
a Adverse reactions were graded according to the Common Terminology Criteria for Adverse Events v5.0. b Consider reverting to previous dose, if appropriate once platelet count recovers to 50,000/mcL or higher. |
||
|
Adverse Reaction |
Severity |
Recommended Dosage Modification |
|
Thrombocytopenia [see Warnings and Precautions (5.3)] |
Platelet count between 25,000/mcL and 50,000/mcL without bleeding |
For patients on 2.5 mg/kg, reduce to Reduced Dosage Level 1 as per Table 1.b For patients on 1.9 mg/kg, continue at same dosage. |
|
Platelet count between 25,000/mcL and 50,000/mcL with bleeding |
Withhold BLENREP until bleeding resolves. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1. For patients on 1.9 mg/kg, resume at same dosage. |
|
|
Platelet count less than 25,000/mcL |
Withhold BLENREP until platelet count recovers to 25,000/mcL or higher. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1. For patients on 1.9 mg/kg, resume at same dosage. |
|
|
Infusion-related Reactions |
Grade 2 |
Interrupt infusion and provide supportive care. Once symptoms resolve to Grade 1 or less, resume infusion at 50% of the initial rate prior to the event. Consider premedication for subsequent infusions. |
|
Grade 3 |
Interrupt infusion and provide supportive care. Once symptoms resolve to Grade 1 or less, resume at 50% of the initial rate prior to the event. Administer premedication for subsequent infusions. |
|
|
Grade 4 |
Permanently discontinue BLENREP. If anaphylactic or life‑threatening infusion reaction, permanently discontinue the infusion and institute appropriate emergency care. |
|
|
Other Adverse Reactions [see Adverse Reactions (6.1] |
Grade 3 |
Withhold BLENREP until adverse reaction improves to Grade 1 or less. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1. For patients on 1.9 mg/kg, resume at same dosage. |
|
Grade 4 |
Consider permanent discontinuation of BLENREP. If continuing treatment, withhold BLENREP until adverse reaction improves to Grade 1 or less. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1. For patients on 1.9 mg/kg, resume at same dosage. |
8.3 Females and Males of Reproductive Potential
BLENREP can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating BLENREP.
Contraception
Females: Advise females of reproductive potential to use effective contraception during BLENREP treatment and for 4 months after the last dose.
Males: Because of the potential for genotoxicity, advise males with female partners of reproductive potential to use effective contraception during treatment with BLENREP and for 6 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings in animal studies, BLENREP may impair fertility in females and males. The effects were not reversible in male rats but were reversible in female rats [see Nonclinical Toxicology (13.1)].
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with belantamab mafodotin‑blmf.
Belantamab mafodotin‑blmf was genotoxic in an in vitro micronucleus assay in human lymphocytes through an aneugenic mechanism. These results are consistent with the pharmacological effect of MMAF binding to tubulin causing microtubule depolymerization resulting in spindle disorganization during cell division. Cys‑mcMMAF was not mutagenic in the bacterial reverse mutation assay (Ames test), the L5178Y mouse lymphoma forward mutation assay, or the in vivo rat bone marrow micronucleus assay.
Fertility studies have not been conducted with belantamab mafodotin‑blmf. Results of repeat‑dose toxicity studies with intravenous administration of belantamab mafodotin‑blmf in rats indicate the potential for impaired male and female reproductive function and fertility. In rats, weekly dosing for 3 weeks at doses ≥10 mg/kg (approximately 5 times the exposure at the maximum recommended human dose [MRHD] of 2.5 mg/kg based on AUC) resulted in degeneration and atrophy of seminiferous tubules in the testes and luteinized nonovulatory follicles in the ovaries. Findings in females were reversible; findings in the testes were not reversible at the end of the 12‑week recovery period with weekly dosing or when given every 3 weeks for 13 weeks at doses ≥10 mg/kg. In male monkeys, the highest dose tested of 10 mg/kg (approximately 5 times the exposure at the MRHD based on AUC) given weekly for 13 weeks resulted in seminiferous tubules degeneration in the testes that was fully reversed following the 12‑week recovery period.
5.2 Blenrep Risk Evaluation and Mitigation Strategy (rems)
BLENREP is available only through a restricted program called the BLENREP REMS because of the risk of ocular toxicity [see Warnings and Precautions (5.1)].
Notable requirements of the BLENREP REMS include the following:
-
•Prescribers must be certified in the BLENREP REMS by enrolling and completing training.
-
•Prescribers must counsel patients receiving BLENREP on the risk of ocular toxicity, the need for monitoring via ophthalmic exams before each dose, and provide patients with the BLENREP REMS Patient Guide.
-
•Patients must be enrolled in the BLENREP REMS and adhere to monitoring.
-
•Healthcare settings that dispense BLENREP must be certified in the BLENREP REMS by enrolling and must obtain authorization prior to dispensing.
-
•Wholesalers and distributors must distribute BLENREP only to certified healthcare settings.
Further information is available at www.BLENREPREMS.com and 1‑855‑690-9572.
Structured Label Content
Section 42231-1 (42231-1)
|
MEDICATION GUIDE BLENREP (BLEN-REP) (belantamab mafodotin-blmf) for injection, for intravenous use |
||
|
What is the most important information I should know about BLENREP? BLENREP can cause serious side effects, including:
|
||
|
|
|
|
Ulcers on the surface of the eye (corneal ulcers), including with infection, may also happen during treatment with BLENREP. Tell your healthcare provider right away if you notice any new or worsening eye symptoms or vision changes during treatment with BLENREP. Your healthcare provider will refer you to an eye care specialist (such as an ophthalmologist or optometrist) to check your eyes before you start treatment, before you receive each dose of BLENREP, and as needed for any new or worsening eye problems. It is important that you:
Because of the risk of eye problems, BLENREP is available only through a restricted program called the BLENREP Risk Evaluation and Mitigation Strategy (REMS). Your healthcare provider will give you the BLENREP REMS Patient Guide, which describes the risk of eye problems, and will explain the REMS program to you. To receive BLENREP, you must enroll in the REMS program and receive eye exams during treatment. See “What are the possible side effects of BLENREP?” for more information about side effects. |
||
|
What is BLENREP? BLENREP is a prescription medicine used in combination with the medicines bortezomib and dexamethasone to treat adults with multiple myeloma who:
It is not known if BLENREP is safe and effective in children. |
||
|
Before receiving BLENREP, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over‑the‑counter medicines, vitamins, and herbal supplements. |
||
|
How will I receive BLENREP?
|
||
|
What are the possible side effects of BLENREP? BLENREP can cause serious side effects, including:
The most common side effects of BLENREP when given with bortezomib and dexamethasone include: |
||
|
|
|
|
The most common severe abnormal blood test results during treatment with BLENREP include decreases in platelets, white blood cells, and hemoglobin, and increases in certain liver enzymes. Tell your healthcare provider right away if you get new or worsening unexplained signs or symptoms of lung problems, including shortness of breath, chest pain, or cough. Your healthcare provider may decrease your dose, temporarily stop, or completely stop treatment with BLENREP if you get serious side effects. BLENREP may affect fertility in males and females, which may affect your ability to have children. Talk to your healthcare provider if this is a concern for you. These are not all of the possible side effects of BLENREP. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‑800‑FDA‑1088. |
||
|
General information about the safe and effective use of BLENREP. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about BLENREP that is written for health professionals. |
||
|
What are the ingredients in BLENREP? Active Ingredient: belantamab mafodotin-blmf Inactive Ingredients: citric acid monohydrate, edetate disodium, polysorbate 80, sodium citrate, and trehalose. |
||
|
Manufactured by: GlaxoSmithKline LLC Philadelphia PA, 19104 U.S. License No. 1727 |
For: GlaxoSmithKline, Durham, NC 27701 ©2025 GSK group of companies or its licensor. BLP:1MG |
|
|
For more information, call GlaxoSmithKline (GSK) at 1‑888‑825‑5249. Trademarks are owned by or licensed to the GSK group of companies. |
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 10/2025
Section 51945-4 (51945-4)
NDC 0173-0913-01
BLENREP
(belantamab mafodotin-blmf)
for injection
70 mg/vial
Rx Only
For intravenous infusion after reconstitution and dilution.
CAUTION: Hazardous Drug.
No preservative.
Dispense the enclosed Medication Guide to each patient.
Contains one Single-Dose vial.
Discard Unused Portion.
2025 GSK group of companies or its licensor.
Product Italy
Rev. 04/25
62000000097965
15 References (15 REFERENCES)
-
1.“OSHA Hazardous Drugs.” OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, BLENREP can cause fetal harm when administered to a pregnant woman, because it contains a genotoxic compound (the microtubule inhibitor, MMAF) and it targets actively dividing cells [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)]. Human immunoglobulin G (IgG) is known to cross the placenta; therefore, belantamab mafodotin-blmf has the potential to be transmitted from the mother to the developing fetus. There are no available data on the use of BLENREP in pregnant women to evaluate for drug-associated risk. No animal reproduction studies were conducted with BLENREP. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data: Animal reproductive or developmental toxicity studies were not conducted with belantamab mafodotin‑blmf. The cytotoxic component of BLENREP, mcMMAF, disrupts microtubule function, is genotoxic, and can be toxic to rapidly dividing cells, suggesting it has the potential to cause embryo‑fetal toxicity.
8.2 Lactation
Risk Summary
There are no data on the presence of belantamab mafodotin-blmf in human milk or the effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with BLENREP and for 3 months after the last dose.
11 Description (11 DESCRIPTION)
Belantamab mafodotin‑blmf is a B‑cell maturation antigen (BCMA)‑directed antibody and microtubule inhibitor conjugate. Belantamab mafodotin‑blmf is an antibody conjugate composed of 3 components: 1) afucosylated, humanized immunoglobulin G1 monoclonal antibody covalently linked to 2) the microtubule inhibitor mcMMAF via 3) a protease‑resistant maleimidocaproyl linker.
The antibody is produced in a mammalian cell line (Chinese Hamster Ovary) using recombinant DNA technology and the microtubule inhibitor and linker are produced by chemical synthesis. Approximately 4 molecules of mafodotin are attached to each antibody molecule. The molecular weight of belantamab mafodotin‑blmf is approximately 152 kDa. Belantamab mafodotin‑blmf has the following structure:
BLENREP (belantamab mafodotin‑blmf) for injection is a sterile, preservative‑free, white to yellow, lyophilized powder in a single‑dose vial for reconstitution and further dilution prior to intravenous use. BLENREP is supplied as 70 mg per vial and requires reconstitution with 1.4 mL of Sterile Water for Injection, USP, to obtain a concentration of 50 mg/mL. Each mL of reconstituted solution contains belantamab mafodotin‑blmf (50 mg) and the inactive ingredients, citric acid monohydrate (0.46 mg), edetate disodium (0.017 mg), polysorbate 80 (0.2 mg), sodium citrate (5.88 mg), and trehalose (68.4 mg). The pH of the reconstituted solution is 6.2.
8.4 Pediatric Use
The safety and effectiveness of BLENREP in pediatric patients have not been established.
8.5 Geriatric Use
Of 242 patients who received BLENREP in DREAMM-7, 121 patients (50%) were aged 65 years or older and 37 patients (15%) were 75 years or older. No overall differences in the safety of BLENREP were observed between patients 65 years of age and older and younger adult patients.
Of the 108 patients who received BLENREP and were evaluated for efficacy in the DREAMM-7 study, there was an insufficient number of older adult patients to determine if the effectiveness in patients 65 years of age and older is different than in younger adult patients.
12.6 Immunogenicity
The observed incidence of anti‑drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti‑drug antibodies in the studies described below with the incidence of anti‑drug antibodies (ADAs) in other studies, including those of BLENREP or of other belantamab mafodotin‑blmf products.
In the combination therapy studies, 3% of patients (15/515) tested positive for treatment‑emergent belantamab mafodotin‑blmf ADAs. Among the 15 patients who tested positive for ADAs, 2 patients tested positive for neutralizing anti‑belantamab mafodotin‑blmf antibodies.
14 Clinical Studies (14 CLINICAL STUDIES)
Relapsed or Refractory Multiple Myeloma in Combination with Bortezomib and Dexamethasone
The efficacy of BLENREP in combination with bortezomib and dexamethasone (BVd) compared with daratumumab, bortezomib, and dexamethasone (DVd) was evaluated in DREAMM‑7 (NCT04246047), an open‑label, randomized, multicenter study in adult patients with relapsed or refractory multiple myeloma. Patients received at least one prior line of therapy and had documented disease progression during or after their most recent line of treatment. Patients who were refractory or intolerant to daratumumab or bortezomib, or with prior exposure to anti‑BCMA therapy were excluded. Patients with existing corneal disease, except for mild punctate keratopathy, were excluded.
Patients were randomized 1:1 to the following treatment arms:
-
•BLENREP 2.5 mg/kg (IV) every 3 weeks on Day 1 of each 21‑day cycle. Bortezomib 1.3 mg/m2 (subcutaneously) on Days 1, 4, 8, and 11 and dexamethasone 20 mg (IV or orally) on the day of, and the day after, bortezomib treatment of Cycles 1–8 (21‑day cycles). From Cycle 9 onward, treatment with BLENREP was continued as a single agent.
-
•Daratumumab 16 mg/kg (IV) every week for Cycles 1–3 and every 3 weeks for Cycles 4–8. Bortezomib 1.3 mg/m2 (subcutaneously) on Days 1, 4, 8, and 11 and dexamethasone 20 mg (IV or orally) on the day of, and the day after, bortezomib treatment of Cycles 1–8 (21‑day cycles). From Cycle 9 onward, treatment with daratumumab was continued as a single agent.
The dose level of dexamethasone in each arm was reduced by half in patients aged 75 years or older. Treatment with BLENREP or daratumumab was continued until disease progression or unacceptable toxicity.
Efficacy was established based on progression-free survival (PFS) and overall survival (OS).
A total of 217 patients who received at least two prior lines of therapy, including a proteasome inhibitor and immunomodulatory agent, were evaluated for efficacy: 108 in the BVd arm and 109 in the DVd arm. Baseline demographics and characteristics were similar across arms. The median age was 65 years (range: 39 to 86); 43% were age 65 to 74 years, 11% age 75 years or older; 53% were male; 86% were White, 11% Asian, 2% Black; R-ISS stage at screening was stage I in 37%, stage II in 56%, stage III in 6%; high‑risk cytogenetics (presence of t (11;14), t (14;16) or 17p13del) were present in 29%; extramedullary disease was present in 11%. The median number of prior lines of therapy was 3 (range: 2 to 7); 2% received a prior anti-CD38 monoclonal antibody, and 71% previously received autologous stem cell transplantation. Overall, 19% of patients were refractory to proteasome inhibitors and 59% were refractory to immunomodulatory agents.
Efficacy results are summarized in Table 8 and Figures 1 and 2.
|
BLENREP +
Bortezomib and Dexamethasone N = 108 |
Daratumumab +
Bortezomib and Dexamethasone N = 109 |
|
|---|---|---|
| NR = not reached. a Based on all randomized patients who received at least two prior lines of therapy, including a proteasome inhibitor and immunomodulatory agent. b Median follow-up of 27.9 months. c Response was based on Independent Review Committee per International Myeloma Working Group criteria. d By Brookmeyer and Crowley method. e Based on stratified Cox regression model. f Median follow-up of 38.7 months. g ORR: sCR+CR+VGPR+PR. h Assessed by next generation sequencing assay (clonoSEQ) at 10‑5 threshold. |
||
|
Progression‑Free Survival (PFS)a,b,c |
||
|
Number (%) of patients with event |
42 (39) |
73 (67) |
|
Median in months (95% CI)d |
31.3 (23.5, NR) |
10.4 (7, 13.4) |
|
Hazard ratio (95% CI)e |
0.31 (0.21, 0.47) |
|
|
Overall Survival (OS)a,f |
||
|
Number (%) of patients with event |
33 (31) |
57 (52) |
|
Median in months (95% CI)d |
NR (NR, NR) |
35.7 (21.1, NR) |
|
Hazard ratio (95% CI)e |
0.49 (0.32, 0.76) |
|
|
Overall Response Rate (ORR)a,b,c,g, % (95% CI) |
81.5 (72.9, 88.3) |
56.9 (47, 66.3) |
|
Stringent complete response (sCR), n (%) |
15 (13.9) |
4 (3.7) |
|
Complete response (CR), n (%) |
19 (17.6) |
5 (4.6) |
|
Very good partial response (VGPR), n (%) |
34 (31.5) |
23 (21.1) |
|
Partial response (PR), n (%) |
20 (18.5) |
30 (27.5) |
|
Minimal Residual Disease (MRD) Negativity Ratea,b,h, n (%) |
25 (23.1) |
3 (2.8) |
|
95% CI |
(15.6, 32.2) |
(0.6, 7.8) |
|
MRD Negativity Rate in Patients with CR or Betterb,h |
||
|
Number of patients with CR or better |
34 |
9 |
|
MRD negativity rate, n (%) |
25 (73.5) |
3 (33.3) |
|
(95% CI) |
(55.6, 87.1) |
(7.5, 70.1) |
Figure 1: Kaplan-Meier Curve for Progression‑Free Survival in DREAMM-7
Figure 2: Kaplan-Meier Curve for Overall Survival in DREAMM-7
In patients who achieved response, the median time to response was 1.43 months (range: 0.7 to 8.4 months) in the BVd arm and 1.03 months (range: 0.7 to 11.1 months) in the DVd arm.
4 Contraindications (4 CONTRAINDICATIONS)
None.
5.1 Ocular Toxicity
BLENREP causes ocular toxicity, defined as changes in the corneal epithelium and changes in BCVA based on ophthalmic exam (including slit lamp exam), or other ocular adverse reactions as defined by the CTCAE [see Adverse Reactions (6.1)].
In DREAMM-7, ocular toxicity occurred in 92% of patients, including Grade 3 or 4 in 77% of patients. The most common ocular toxicities (>25%) were reduction in BCVA (89%) and corneal exam findings (86%) based on ophthalmic exam findings, blurred vision (66%), dry eye (51%), photophobia (47%), foreign body sensation in eyes (44%), eye irritation (43%), and eye pain (33%) [see Adverse Reactions (6.1)].
Ocular toxicity based on ophthalmic exam findings was reported as Grade 2 in 9% of patients, Grade 3 in 56% of patients, and Grade 4 in 21% of patients. The median time to onset of the first Grade 2 to 4 ophthalmic exam findings was 43 days (range: 15 to 611 days). The median duration of all Grade 2 to 4 ophthalmic exam findings was 85 days (range: 5 to 813 days). Patients experienced a median of 3 episodes (range: 1 to 11 episodes) of ocular toxicity based on ophthalmic exam findings. Of the patients with Grade 2 to 4 ophthalmic exam findings, 42% had improvement of the last event to Grade 1 or better; 22% had resolution of the last event based on return to baseline or normal ophthalmic exam findings.
The most commonly reported corneal exam findings included superficial punctate keratopathy, microcyst-like deposits, epithelial changes, and haze. Cases of corneal ulcer, including cases with infection, have been reported and should be managed promptly by an eye care professional [see Adverse Reactions (6.1)].
A reduction in BCVA to 20/50 or worse in at least one eye occurred in 69% of patients, including 29% who experienced a change in BCVA to 20/100 or worse, and 12% who experienced a change in BCVA to 20/200 or worse. Of the patients with reduced BCVA to 20/50 or worse in at least one eye, 61% had resolution of the last event to baseline or better. Of the patients with reduced BCVA to 20/100 or worse, 57% had resolution of the last event. Of the patients with reduced BCVA to 20/200 or worse, 48% had resolution of the last event.
Ophthalmic exams (including slit lamp exam and BCVA assessment) should be conducted by an eye care professional, such as an ophthalmologist or optometrist, at baseline, before each dose of BLENREP, promptly for new or worsening symptoms, and as clinically indicated. Perform baseline exam within 4 weeks prior to the first dose. Perform each follow-up exam within 10 days prior to the next planned dose. All effort should be made to schedule the exam as close to BLENREP dosing as possible. Withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less and resume at same or reduced dose or permanently discontinue based on severity [see Dosage and Administration (2.1, 2.3)].
Counsel patients to promptly inform their healthcare provider of any ocular symptoms. Counsel patients to use preservative‑free artificial tears at least 4 times a day starting with the first infusion and continuing until the end of treatment, and to avoid wearing contact lenses for the duration of therapy. Bandage contact lenses may be used under the direction of an eye care professional [see Dosage and Administration (2.1)].
Changes in visual acuity may be associated with difficulty for driving and reading. Counsel patients to use caution when driving or operating machinery.
BLENREP is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) [see Warnings and Precautions (5.2)].
6 Adverse Reactions (6 ADVERSE REACTIONS)
5.3 Thrombocytopenia
Thrombocytopenia of any grade occurred in 100% of patients in DREAMM‑7 [see Adverse Reactions (6.1)].
Grade 2 thrombocytopenia occurred in 10% of patients, Grade 3 in 29% of patients, and Grade 4 in 45% of patients. Clinically significant bleeding (Grade ≥2) occurred in 7% of patients with concomitant low platelet levels (Grade 3 or 4).
Monitor complete blood cell counts at baseline and periodically during treatment as clinically indicated. Withhold or reduce the dose of BLENREP based on severity [see Dosage and Administration (2.3)].
12.2 Pharmacodynamics
Exposure‑Response Relationships
When BLENREP was used in combination with Vd, higher belantamab mafodotin‑blmf Cycle 1 exposure was associated with a higher incidence of some safety adverse reactions (e.g., Grade ≥2 corneal exam findings).
Cardiac Electrophysiology
Belantamab mafodotin‑blmf had no meaningful QTc prolongation (>10 ms) at the recommended dosage.
12.3 Pharmacokinetics
Belantamab mafodotin‑blmf exhibited dose‑proportional pharmacokinetics, with a gradual decrease in clearance over time. After a planned infusion duration of 30 minutes, maximum belantamab mafodotin‑blmf plasma concentrations occurred at or shortly after the end of the infusion. Accumulation of belantamab mafodotin‑blmf was minimal to moderate for the ADC (Cycle 3 to Cycle 1 ratio was 1.13 for Cmax and 1.58 for AUC) as observed with a dosing regimen of every 3 weeks.
Table 7 describes the pharmacokinetics of belantamab mafodotin‑blmf for the 2.5 mg/kg dose on Cycle 1 at the end of the first 3‑week intervals.
| AUCb | Cmax | Ctau | |
|---|---|---|---|
| ADC = antibody drug conjugate; AUC = area under the curve; Cmax = maximum plasma concentration; Ctau = concentration at the end of a dosing interval. a Data presented as geometric mean (CV%). b AUC for ADC is AUC(0‑21days) and for cys-mcMMAF is AUC(0‑7days). |
|||
|
ADC (%) |
3,950 mcg•h/mL (30.6) |
43.7 mcg/mL (22.1) |
2.03 mcg/mL (62.5) |
|
cys‑mcMMAF (%) |
94.2 ng•h/mL (42.3) |
0.976 ng/mL (45.3) |
– |
Distribution
In vitro, cys‑mcMMAF exhibited low protein binding (70% unbound at a concentration of 5 ng/mL) in human plasma.
The geometric mean (CV%) steady‑state volume of distribution of belantamab mafodotin‑blmf was 10.8 L (22.2%).
Elimination
The geometric mean (CV%) belantamab mafodotin‑blmf (ADC) initial systemic clearance (CL) was 0.901 L/day (40%), and the elimination half‑life was 13 days (26%). Following treatment, steady‑state CL was 0.605 L/day (43%) or approximately 33% lower than initial systemic CL with an elimination half‑life of 17 days (31%).
The fraction of intact cys‑mcMMAF excreted in urine was approximately 18% of the dose in Cycle 1, with no evidence of other mcMMAF‑related metabolites.
Metabolism: The monoclonal antibody portion of belantamab mafodotin‑blmf is expected to undergo proteolysis to small peptides and individual amino acids by ubiquitous proteolytic enzymes. Cys‑mcMMAF had limited metabolic clearance in human hepatic S9 fraction incubation studies.
Specific Populations
No clinically significant differences in the pharmacokinetics of belantamab mafodotin‑blmf were observed based on age (32 to 89 years), sex, race (White vs. Black vs. Asian), body weight (37 to 170 kg), mild to severe renal impairment and kidney failure (eGFR <30 mL/min), or mild hepatic impairment (total bilirubin >ULN to ≤1.5 × ULN and any AST or total bilirubin ≤ULN with AST >ULN).
The effects of moderate (total bilirubin >1.5 × ULN to ≤3 × ULN and any AST) or severe hepatic impairment (total bilirubin >3 × ULN and any AST) on the pharmacokinetics of belantamab mafodotin‑blmf are unknown.
In Vitro Studies:
Cytochrome P450 (CYP) Enzymes: Cys-mcMMAF is not an inhibitor, an inducer, or a sensitive substrate of cytochrome P450 enzymes.
Transporter Systems: Cys‑mcMMAF is a substrate of organic anion transporting polypeptide (OATP)1B1 and OATP1B3, multidrug resistance‑associated protein (MRP)1, MRP2, and MRP3, bile salt export pump (BSEP), and a possible substrate of P‑glycoprotein (P‑gp).
2.2 Recommended Dosage
The recommended dosage for BLENREP is 2.5 mg/kg of actual body weight once every 3 weeks in combination with bortezomib and dexamethasone (BVd) for the first 8 cycles, followed by BLENREP 2.5 mg/kg of actual body weight once every 3 weeks as a single agent until disease progression or unacceptable toxicity.
For dosing instructions of agents administered in combination with BLENREP, see Clinical Studies (14) and respective Prescribing Information, as appropriate.
BLENREP is administered as an intravenous infusion over approximately 30 minutes.
8.6 Hepatic Impairment
No dose adjustment is recommended for patients with mild hepatic impairment (total bilirubin > upper limit of normal [ULN] to ≤1.5 × ULN and any aspartate aminotransferase [AST] or total bilirubin ≤ULN with AST >ULN).
The recommended dose of BLENREP has not been established in patients with moderate or severe hepatic impairment [see Clinical Pharmacology (12.3)].
1 Indications and Usage (1 INDICATIONS AND USAGE)
BLENREP is indicated in combination with bortezomib and dexamethasone for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least two prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent.
12.1 Mechanism of Action
Belantamab mafodotin‑blmf is an antibody‑drug conjugate (ADC). The antibody component is an afucosylated IgG1 directed against BCMA, a protein expressed on normal B lymphocytes and multiple myeloma cells. The small molecule component is mcMMAF, a microtubule inhibitor. Upon binding to BCMA, belantamab mafodotin‑blmf is internalized followed by release of the cytotoxic agent (cys‑mcMMAF) via proteolytic cleavage. The released cys‑mcMMAF intracellularly disrupts the microtubule network, leading to cell cycle arrest and apoptosis.
Belantamab mafodotin‑blmf had antitumor activity in multiple myeloma cells and mediated killing of tumor cells through cys‑mcMMAF‑induced apoptosis, as well as by tumor cell lysis through antibody‑dependent cellular cytotoxicity and antibody‑dependent cellular phagocytosis.
Warning: Ocular Toxicity (WARNING: OCULAR TOXICITY)
-
•BLENREP causes changes in the corneal epithelium resulting in changes in vision, including severe visual impairment, and symptoms such as blurred vision and dry eyes. In the clinical study, corneal ulcers, including cases with infection, also occurred [see Warnings and Precautions (5.1)].
-
•Conduct ophthalmic exams at baseline, before each dose, promptly for new or worsening symptoms, and as clinically indicated. In the clinical study, 83% of patients required a dosage modification due to ocular toxicity. Withhold BLENREP until improvement and resume or permanently discontinue, based on severity [see Dosage and Administration (2.3), Warnings and Precautions (5.1)].
-
•Because of the risk of ocular toxicity, BLENREP is available only through a restricted program called the BLENREP Risk Evaluation and Mitigation Strategy (REMS) [see Warnings and Precautions (5.2)].
5.4 Embryo Fetal Toxicity (5.4 Embryo-fetal Toxicity)
Based on its mechanism of action, BLENREP can cause fetal harm when administered to a pregnant woman because it contains a genotoxic compound (the microtubule inhibitor, monomethyl auristatin F [MMAF]) and it targets actively dividing cells.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with BLENREP and for 4 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with BLENREP and for 6 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
-
•Thrombocytopenia: Monitor complete blood counts at baseline and periodically during treatment. Withhold or reduce the dosage based on severity. (2.3, 5.3)
-
•Embryo‑fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to fetus and to use effective contraception. (5.4, 8.1, 8.3)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
-
•The recommended dosage of BLENREP, in combination with bortezomib and dexamethasone, is 2.5 mg/kg as an intravenous infusion over 30 minutes once every 3 weeks for 8 cycles, followed by BLENREP 2.5 mg/kg every 3 weeks as a single agent. (2.2)
-
•See Full Prescribing Information for instructions on preparation and administration. (2.4)
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
For injection: 70 mg of belantamab mafodotin-blmf as a white to yellow lyophilized powder in a single-dose vial for reconstitution and further dilution.
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
Lactation: Advise not to breastfeed. (8.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Relapsed or Refractory Multiple Myeloma in Combination with Bortezomib and Dexamethasone
The safety of BLENREP with bortezomib and dexamethasone (n = 242) compared with daratumumab with bortezomib and dexamethasone (n = 246) was evaluated in DREAMM‑7 in patients with relapsed or refractory multiple myeloma who received at least one prior line of therapy [see Clinical Studies (14)]. Patients received BLENREP 2.5 mg/kg of actual body weight once every 3 weeks in combination with bortezomib and dexamethasone (BVd) for the first 8 cycles, followed by BLENREP as a single agent or daratumumab in combination with bortezomib and dexamethasone (DVd) for the first 8 cycles, followed by daratumumab as a single agent. Among patients who received BLENREP, 69% were exposed for 6 months or longer and 55% were exposed for greater than one year. The safety of BLENREP in combination with bortezomib and dexamethasone in patients who received only one prior line of therapy (n = 125) has not been established.
Serious adverse reactions occurred in 50% of patients who received BVd. Serious adverse reactions in ≥2% of patients included pneumonia (18%), pyrexia (5%), thrombocytopenia (5%), COVID-19 (5%), upper respiratory tract infection (4%), sepsis (4%), second primary malignancy (3%), and anemia (2%). Fatal adverse reactions occurred in 10% of patients who received BVd. Fatal adverse reactions which occurred in >1 patient included pneumonia (4%), sepsis (2%), COVID-19 (1%), respiratory failure (<1%), and intracranial hemorrhage (<1%).
Permanent discontinuation of BLENREP due to an adverse reaction occurred in 17% of patients. Adverse reactions which resulted in permanent discontinuation of BLENREP in ≥3% of patients included ocular toxicity (9%) and pneumonia (4%).
Dosage interruptions of BLENREP due to an adverse reaction occurred in 78% of patients. Adverse reactions which required dosage interruption of BLENREP in ≥3% of patients included ocular toxicity based on ophthalmic exam findings (74%), blurred vision (32%), upper respiratory tract infection (20%), dry eye (14%), photophobia (14%), pneumonia (14%), eye irritation (13%), COVID-19 (12%), foreign body sensation in eyes (12%), eye pain (10%), thrombocytopenia (9%), visual impairment (7%), cataract (5%), diarrhea (4%), and neutropenia (4%).
Dosage reductions of BLENREP due to an adverse reaction occurred in 36% of patients. Adverse reactions which required dosage reductions for BLENREP in ≥3% of patients included ocular toxicity based on ophthalmic exam findings (30%), thrombocytopenia (14%), and blurred vision (10%).
The most common adverse reactions (≥20%) were reduction in BCVA, corneal exam findings, blurred vision, dry eye, photophobia, foreign body sensation in eyes, eye irritation, upper respiratory tract infection, hepatotoxicity, eye pain, diarrhea, fatigue, pneumonia, cataract, and COVID-19. The most common Grade 3 or 4 laboratory abnormalities (≥10%) were decreased platelets, decreased lymphocytes, decreased neutrophils, increased gamma glutamyltransferase, decreased white blood cells, and decreased hemoglobin.
Table 4 summarizes the adverse reactions in DREAMM‑7.
| BCVA = best‑corrected visual acuity; BVd = BLENREP + bortezomib and dexamethasone; DVd = daratumumab + bortezomib and dexamethasone. a Adverse reactions, except ophthalmic exam findings, were graded according to Common Terminology Criteria for Adverse Events v5.0. b Based on ophthalmic exam findings. c Grouped term includes other related terms. d Includes the following fatal adverse reactions: BVd: pneumonia (n = 10), COVID-19 (n = 3), upper respiratory tract infection (n = 1); DVd: pneumonia (n = 7), COVID-19 (n = 5), pyrexia (n = 1). |
||||
|
Adverse Reactiona |
BLENREP + Bortezomib and Dexamethasone N = 242 |
Daratumumab + Bortezomib and Dexamethasone N = 246 |
||
|
All Grades (%) |
Grades 3‑4 (%) |
All Grades (%) |
Grades 3‑4 (%) |
|
|
Eye disorders |
||||
|
Reduction in BCVAb |
89 |
57 |
44 |
9 |
|
Corneal exam findingsb |
86 |
72 |
19 |
3 |
|
Blurred vision |
66 |
22 |
11 |
0.8 |
|
Dry eyec |
51 |
7 |
7 |
0 |
|
Photophobia |
47 |
2 |
2 |
0 |
|
Foreign body sensation in eyesc |
44 |
3 |
4 |
0 |
|
Eye irritation |
43 |
5 |
5 |
0 |
|
Eye painc |
33 |
0.8 |
4 |
0.4 |
|
Cataractc |
24 |
8 |
14 |
3 |
|
Visual impairment |
11 |
5 |
2 |
0.4 |
|
Gastrointestinal disorders |
||||
|
Diarrhea |
32 |
4 |
31 |
4 |
|
Nausea |
16 |
0.8 |
12 |
0 |
|
Infections |
||||
|
Upper respiratory tract infectionc,d |
38 |
2 |
36 |
2 |
|
Pneumoniac,d |
26 |
16 |
17 |
5 |
|
COVID-19d |
24 |
5 |
20 |
2 |
|
Hepatobiliary disorders |
||||
|
Hepatotoxicityc |
33 |
14 |
16 |
2 |
|
General disorders and administration site conditions |
||||
|
Fatiguec |
26 |
6 |
27 |
4 |
|
Pyrexiac,d |
19 |
0.4 |
11 |
2 |
Clinically relevant adverse reactions in <10% of patients who received BVd included: increased lacrimation, vomiting, diplopia, albuminuria, sepsis, eye pruritus, infusion-related reactions, corneal ulcer (including cases with infection), and pneumonitis.
Table 5 summarizes the laboratory abnormalities in DREAMM-7.
| Laboratory Abnormality |
BLENREP +
Bortezomib and Dexamethasonea |
Daratumumab +
Bortezomib and Dexamethasonea |
||
|---|---|---|---|---|
|
All Grades
(%) |
Grades 3‑4
(%) |
All Grades
(%) |
Grades 3‑4
(%) |
|
| BVd = BLENREP + bortezomib and dexamethasone; DVd = daratumumab + bortezomib and dexamethasone. a The denominator used to calculate the rate varied from 238 to 241 (BVd) and 243 to 246 (DVd) based on the number of patients with a baseline value and at least one post-treatment value. |
||||
|
Hematology |
||||
|
Platelets decreased |
100 |
74 |
88 |
48 |
|
Lymphocytes decreased |
90 |
53 |
92 |
56 |
|
Leukocytes decreased |
59 |
11 |
67 |
17 |
|
Neutrophils decreased |
52 |
17 |
53 |
13 |
|
Hemoglobin decreased |
51 |
10 |
60 |
12 |
|
Chemistry |
||||
|
Aspartate aminotransferase increased |
88 |
5 |
40 |
0 |
|
Gamma glutamyltransferase increased |
73 |
15 |
44 |
3 |
|
Alanine aminotransferase increased |
71 |
5 |
54 |
1 |
|
Creatinine increased |
51 |
2 |
53 |
<1 |
|
Creatine phosphokinase increased |
48 |
3 |
31 |
3 |
Patient-reported ocular symptoms were assessed using the Patient-Reported Outcomes Common Terminology Criteria for Adverse Events (PRO-CTCAE) and Ocular Surface Disease Index (OSDI).
PRO-CTCAE assessments were collected at baseline and then every 3 weeks until treatment discontinuation. Completion rates in both arms were ≥90% at baseline and ≥81% at subsequent timepoints where >50% of patients remained on treatment. OSDI assessments were collected every 3 weeks until the 6th dose and then every 6 weeks thereafter until treatment discontinuation for BVd and every 3 weeks until Cycle 6 and then every 12 weeks thereafter until treatment discontinuation for DVd. Completion rates in both arms were ≥90% at baseline and ≥52% at subsequent timepoints where >50% of patients remained on treatment.
Table 6 summarizes the patient-reported symptom of blurred vision as assessed by PRO-CTCAE.
| Symptom (Attribute)a | Any Symptom Before Treatmentb | Score 3 or 4 Before Treatmentc | Any Symptom on Treatmentd,e | Score 3 or 4 on Treatmentd,f | ||||
|---|---|---|---|---|---|---|---|---|
|
BVd (%)
n = 232 |
DVd (%)
n = 227 |
BVd (%)
n = 232 |
DVd (%)
n = 227 |
BVd (%)
n = 238 |
DVd (%)
n = 239 |
BVd (%)
n = 238 |
DVd (%)
n = 239 |
|
| BVd = BLENREP + bortezomib and dexamethasone; DVd = daratumumab + bortezomib and dexamethasone; PRO-CTCAE = patient-reported outcomes common terminology criteria for adverse events. a Symptom attribute scoring defined as severity with a score of 0 = ‘none’; 1 = ‘mild’; 2 = ‘moderate’; 3 = ‘severe’; 4 = ‘very severe’. b Percentage of patients whose symptom score before treatment was 1 to 4. c Percentage of patients whose symptom score before treatment was 3 or 4. d Number of patients who provided at least one on-treatment score. e Percentage of patients whose maximum post-baseline score was 1 to 4 on treatment. f Percentage of patients whose maximum post-baseline score was 3 or 4 on treatment. |
||||||||
|
Blurred vision (severity) |
28 |
24 |
<1 |
2 |
93 |
74 |
55 |
15 |
Driving at night was assessed using the OSDI. At baseline, the proportion of patients who reported limitations with driving at night “all of the time” or “most of the time” during the last week was 9% in the BVd arm and 4% in the DVd arm. The proportion of patients who reported limitations with driving at night “all of the time” or “most of the time” during the last week was highest in the BVd arm at 47% at Week 13 and in the DVd arm at 12% at Week 76.
2.1 Important Safety Information
Ophthalmic exams, including slit lamp exam and assessment of best‑corrected visual acuity (BCVA), should be conducted by an eye care professional, such as an ophthalmologist or optometrist. Conduct ophthalmic exams at baseline, before each dose of BLENREP, promptly for new or worsening symptoms, and as clinically indicated [see Warnings and Precautions (5.1)].
Counsel patients to promptly inform their healthcare provider of any ocular symptoms. Advise patients to use preservative‑free artificial tears at least 4 times a day starting with the first infusion and continuing until end of treatment, and to avoid wearing contact lenses for the duration of therapy. Bandage contact lenses may be used under the direction of an eye care professional [see Warnings and Precautions (5.1)].
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA‑approved patient labeling (Medication Guide).
Ocular Toxicity
-
•Advise patients that ocular toxicity may occur during treatment with BLENREP [see Warnings and Precautions (5.1)].
-
•Advise patients to promptly tell their healthcare provider if they notice any new or worsening eye symptoms [see Dosage and Administration (2.1), Warnings and Precautions (5.1)].
-
•Advise patients that they will be sent to an eye care professional to obtain ophthalmic exams before starting BLENREP, before each dose, promptly for any new or worsening eye symptoms, and as clinically indicated [see Dosage and Administration (2.1), Warnings and Precautions (5.1)].
-
•Advise patients to administer preservative‑free artificial tears at least 4 times per day starting with the first infusion and continuing until the end of treatment and to avoid wearing contact lenses for the duration of therapy. Bandage contact lenses may be used under the direction of an eye care professional [see Dosage and Administration (2.1, 2.3), Warnings and Precautions (5.1)].
-
•Advise patients to use caution when driving or operating machinery as BLENREP may adversely affect their vision [see Warnings and Precautions (5.1)].
BLENREP REMS
-
•Advise patients that because of the risk of ocular toxicity, BLENREP is available only through a restricted program called the BLENREP REMS [see Warnings and Precautions (5.2)].
-
•Patients must receive counseling about the risk of ocular toxicity, enroll in the REMS, and adhere to ongoing monitoring via ophthalmic exams. Patients will be given the BLENREP REMS Patient Guide. This guide describes the risk of ocular toxicity with BLENREP [see Warnings and Precautions (5.2)].
Thrombocytopenia
-
•Advise patients to inform their healthcare provider if they develop signs or symptoms of bleeding [see Warnings and Precautions (5.3)].
Embryo‑fetal Toxicity
-
•Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4), Use in Specific Populations (8.1, 8.3)].
-
•Advise females of reproductive potential to use effective contraception during treatment with BLENREP and for 4 months after the last dose [see Warnings and Precautions (5.4), Use in Specific Populations (8.3)].
-
•Advise males with female partners of reproductive potential to use effective contraception during treatment with BLENREP and for 6 months after the last dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
-
•Advise women not to breastfeed during treatment with BLENREP and for 3 months after the last dose [see Use in Specific Populations (8.2)].
Infertility
-
•Advise males and females of reproductive potential that BLENREP may impair fertility [see Use in Specific Populations (8.3)].
Pneumonitis
-
•Advise patients to immediately report any new or worsening respiratory symptoms to their healthcare provider [see Adverse Reactions (6.1)].
Trademarks are owned by or licensed to the GSK group of companies.
Manufactured by:
GlaxoSmithKline LLC
Philadelphia PA, 19104
U.S. License No. 1727
For:
GlaxoSmithKline
Durham, NC 27701
©2025 GSK group of companies or its licensor.
BLP:1PI
2.4 Preparation and Administration
BLENREP is a hazardous drug. Follow applicable special handling and disposal procedures.1
Calculate the dose (mg), total volume (mL) of solution required, and the number of vials of BLENREP needed based on the patient’s actual body weight. More than one vial may be needed for a full dose.
Reconstitution
-
•Remove the vial(s) of BLENREP from the refrigerator and allow to stand for approximately 10 minutes to reach room temperature (68°F to 77°F [20°C to 25°C]).
-
•Reconstitute each 70 mg vial of BLENREP with 1.4 mL of Sterile Water for Injection, USP, to obtain a final concentration of 50 mg/mL. Gently swirl the vial to aid dissolution. Do not shake.
-
•If the reconstituted solution is not used immediately, store in the original container refrigerated at 36ºF to 46ºF (2ºC to 8ºC) or at room temperature (68°F to 77°F [20°C to 25°C]) for up to 4 hours. Discard if not diluted within 4 hours. Do not freeze.
-
•Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The reconstituted solution should be a clear to opalescent, colorless to yellow to brown liquid. Discard if extraneous particulate matter is observed.
Dilution
-
•Withdraw the calculated volume of BLENREP from the appropriate number of vials and dilute in a 250 mL infusion bag of 0.9% Sodium Chloride Injection, USP, to a final concentration of 0.2 mg/mL to 2 mg/mL. The infusion bag must be made of polyvinylchloride (PVC) or polyolefin (PO).
-
•Mix the diluted solution by gentle inversion. Do not shake.
-
•Discard any unused reconstituted solution of BLENREP left in the vial(s).
-
•If the diluted infusion solution is not used immediately, store refrigerated at 36ºF to 46ºF (2ºC to 8ºC) for up to 24 hours. Do not freeze. Once removed from refrigeration, administer the diluted infusion solution of BLENREP within 6 hours (including infusion time).
-
•Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The diluted infusion solution should be clear and colorless. Discard if particulate matter is observed.
Administration
-
•If refrigerated, allow the diluted infusion solution to equilibrate to room temperature (68ºF to 77ºF [20ºC to 25ºC]) prior to administration. Diluted infusion solution may be kept at room temperature for no more than 6 hours (including infusion time).
-
•Administer by intravenous infusion over approximately 30 minutes using an infusion set made of PVC or PO.
-
•Filtration of the diluted solution is not required; however, if the diluted solution is filtered, use a polyethersulfone (PES)-based filter (0.2 micron).
Do not mix or administer BLENREP with other products. The product does not contain a preservative.
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
BLENREP (belantamab mafodotin‑blmf) for injection is a sterile, preservative‑free, white to yellow lyophilized powder for reconstitution and further dilution prior to intravenous use.
BLENREP is supplied in a carton containing one 70 mg single‑dose vial with a rubber stopper (not made with natural rubber latex) and aluminum overseal with removable cap (NDC 0173‑0913‑01).
Store vials refrigerated at 36ºF to 46ºF (2ºC to 8ºC).
BLENREP is a hazardous drug. Follow applicable special handling and disposal procedures.1
2.3 Dosage Modifications for Adverse Reactions
In the BVd arm of the clinical study, 98% of patients required a dosage modification for any component of treatment for an adverse reaction, including 87% who required a dosage modification of BLENREP [see Clinical Studies (14)]. Eighty-three percent of patients required a dosage modification of BLENREP for ocular toxicity based on ophthalmic exam findings or other ocular adverse reactions as defined by the Common Terminology Criteria for Adverse Events (CTCAE) [see Adverse Reactions (6.1)]. There were high rates of dosage modifications in early treatment cycles. By Cycle 3, 53% of patients had a dosage interruption or reduction, 7% had discontinued treatment, and only 40% received the planned dose of BLENREP.
The recommended dosage modifications for ocular toxicity based on ophthalmic exam findings are provided in Tables 1 and 2. Ophthalmic exam findings include both corneal exam findings and change in BCVA as assessed by an eye care professional. The overall grade of ophthalmic exam findings is based on the worst finding in the worst affected eye, based on either corneal exam finding or a change in BCVA. Corneal exam findings may or may not be accompanied by changes in BCVA or ocular symptoms.
-
•Do not re‑escalate the dose of BLENREP after a dosage reduction is made for ocular toxicity based on ophthalmic exam findings.
-
•In the clinical study, 67% of patients required a dosage interruption of BLENREP for ocular toxicity that lasted longer than 3 weeks (time between doses, median: 5.7 weeks [range: 3 to 31 weeks]).
The recommended dosage modifications for other adverse reactions, including dosage modifications for ocular adverse reactions based on the CTCAE, are provided in Table 3.
| a Reduced Dosage Level 2 is specific to dosage reductions due to ocular toxicity based on ophthalmic exam findings. | |
|
BLENREP |
|
|
Reduced Dosage Level 1 |
1.9 mg/kg every 3 weeks |
|
Reduced Dosage Level 2a |
1.9 mg/kg every 8 weeks |
| BCVA = best‑corrected visual acuity. a Mild superficial keratopathy (documented worsening from baseline). Refer to Table 3 for recommended dosage modifications for other ocular adverse reactions. b Microcyst‑like deposits are considered at least a Grade 2 finding. Withhold BLENREP if any microcyst‑like deposits are observed. |
||
|
Severity |
Ophthalmic Exam Findings [see Warnings and Precautions (5.1)] |
Recommended Dosage Modification |
|
Grade 1 |
Corneal Exam Findings:
and/or Change in BCVA:
|
Continue treatment at current dosage. |
|
Grade 2 |
Corneal Exam Findings:
and/or Change in BCVA:
|
Withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less. Resume treatment at Reduced Dosage Level 1 as per Table 1. If recurrent Grade 2 or 3 ocular toxicity is experienced, resume treatment at Reduced Dosage Level 2. |
|
Grade 3 |
Corneal Exam Findings:
and/or Change in BCVA:
|
|
|
Grade 4 |
Corneal Exam Findings:
and/or Change in BCVA:
|
Consider permanent discontinuation of BLENREP. If continuing treatment, withhold BLENREP until improvement in both corneal exam findings and change in BCVA to Grade 1 or less. For patients previously on 2.5 mg/kg every 3 weeks, resume treatment at Reduced Dosage Level 1 as per Table 1. For patients previously on 1.9 mg/kg every 3 weeks, resume treatment at Reduced Dosage Level 2. If recurrent Grade 4 ocular toxicity is experienced, permanently discontinue BLENREP. |
|
a Adverse reactions were graded according to the Common Terminology Criteria for Adverse Events v5.0. b Consider reverting to previous dose, if appropriate once platelet count recovers to 50,000/mcL or higher. |
||
|
Adverse Reaction |
Severity |
Recommended Dosage Modification |
|
Thrombocytopenia [see Warnings and Precautions (5.3)] |
Platelet count between 25,000/mcL and 50,000/mcL without bleeding |
For patients on 2.5 mg/kg, reduce to Reduced Dosage Level 1 as per Table 1.b For patients on 1.9 mg/kg, continue at same dosage. |
|
Platelet count between 25,000/mcL and 50,000/mcL with bleeding |
Withhold BLENREP until bleeding resolves. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1. For patients on 1.9 mg/kg, resume at same dosage. |
|
|
Platelet count less than 25,000/mcL |
Withhold BLENREP until platelet count recovers to 25,000/mcL or higher. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1. For patients on 1.9 mg/kg, resume at same dosage. |
|
|
Infusion-related Reactions |
Grade 2 |
Interrupt infusion and provide supportive care. Once symptoms resolve to Grade 1 or less, resume infusion at 50% of the initial rate prior to the event. Consider premedication for subsequent infusions. |
|
Grade 3 |
Interrupt infusion and provide supportive care. Once symptoms resolve to Grade 1 or less, resume at 50% of the initial rate prior to the event. Administer premedication for subsequent infusions. |
|
|
Grade 4 |
Permanently discontinue BLENREP. If anaphylactic or life‑threatening infusion reaction, permanently discontinue the infusion and institute appropriate emergency care. |
|
|
Other Adverse Reactions [see Adverse Reactions (6.1] |
Grade 3 |
Withhold BLENREP until adverse reaction improves to Grade 1 or less. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1. For patients on 1.9 mg/kg, resume at same dosage. |
|
Grade 4 |
Consider permanent discontinuation of BLENREP. If continuing treatment, withhold BLENREP until adverse reaction improves to Grade 1 or less. For patients previously on 2.5 mg/kg, resume at Reduced Dosage Level 1 as per Table 1. For patients on 1.9 mg/kg, resume at same dosage. |
8.3 Females and Males of Reproductive Potential
BLENREP can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating BLENREP.
Contraception
Females: Advise females of reproductive potential to use effective contraception during BLENREP treatment and for 4 months after the last dose.
Males: Because of the potential for genotoxicity, advise males with female partners of reproductive potential to use effective contraception during treatment with BLENREP and for 6 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings in animal studies, BLENREP may impair fertility in females and males. The effects were not reversible in male rats but were reversible in female rats [see Nonclinical Toxicology (13.1)].
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with belantamab mafodotin‑blmf.
Belantamab mafodotin‑blmf was genotoxic in an in vitro micronucleus assay in human lymphocytes through an aneugenic mechanism. These results are consistent with the pharmacological effect of MMAF binding to tubulin causing microtubule depolymerization resulting in spindle disorganization during cell division. Cys‑mcMMAF was not mutagenic in the bacterial reverse mutation assay (Ames test), the L5178Y mouse lymphoma forward mutation assay, or the in vivo rat bone marrow micronucleus assay.
Fertility studies have not been conducted with belantamab mafodotin‑blmf. Results of repeat‑dose toxicity studies with intravenous administration of belantamab mafodotin‑blmf in rats indicate the potential for impaired male and female reproductive function and fertility. In rats, weekly dosing for 3 weeks at doses ≥10 mg/kg (approximately 5 times the exposure at the maximum recommended human dose [MRHD] of 2.5 mg/kg based on AUC) resulted in degeneration and atrophy of seminiferous tubules in the testes and luteinized nonovulatory follicles in the ovaries. Findings in females were reversible; findings in the testes were not reversible at the end of the 12‑week recovery period with weekly dosing or when given every 3 weeks for 13 weeks at doses ≥10 mg/kg. In male monkeys, the highest dose tested of 10 mg/kg (approximately 5 times the exposure at the MRHD based on AUC) given weekly for 13 weeks resulted in seminiferous tubules degeneration in the testes that was fully reversed following the 12‑week recovery period.
5.2 Blenrep Risk Evaluation and Mitigation Strategy (rems) (5.2 BLENREP Risk Evaluation and Mitigation Strategy (REMS))
BLENREP is available only through a restricted program called the BLENREP REMS because of the risk of ocular toxicity [see Warnings and Precautions (5.1)].
Notable requirements of the BLENREP REMS include the following:
-
•Prescribers must be certified in the BLENREP REMS by enrolling and completing training.
-
•Prescribers must counsel patients receiving BLENREP on the risk of ocular toxicity, the need for monitoring via ophthalmic exams before each dose, and provide patients with the BLENREP REMS Patient Guide.
-
•Patients must be enrolled in the BLENREP REMS and adhere to monitoring.
-
•Healthcare settings that dispense BLENREP must be certified in the BLENREP REMS by enrolling and must obtain authorization prior to dispensing.
-
•Wholesalers and distributors must distribute BLENREP only to certified healthcare settings.
Further information is available at www.BLENREPREMS.com and 1‑855‑690-9572.
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:54:30.041013 · Updated: 2026-03-14T22:49:04.413893