Pralatrexate Injection
a5daa583-4965-4564-ad87-41ffe6c08a11
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
Pralatrexate injection is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL). This indication is approved under accelerated approval based on overall response rate [see Clinical Studies ( 14 )] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Dosage and Administration
Supplement patients with vitamin B 12 mg intramuscularly every 8-10 weeks starting 10 weeks before the first dose and folic acid 1 to 1.25 mg orally once daily starting 10 days before the first dose. ( 2.1 ) The recommended dosage of Pralatrexate injection is 30 mg/m 2 intravenously over 3 to 5 minutes once weekly for 6 weeks in 7-week cycles. ( 2.1 ) For patients with severe renal impairment (GFR 15 to 29 mL/min/1.73 m 2 ), reduce the Pralatrexate injection dose to 15 mg/m 2 ( 2.1 ).
Contraindications
None
Warnings and Precautions
Myelosuppression : Monitor complete blood counts and omit and/or reduce dose based on ANC and platelet count. ( 2.4 , 5.1 ) Mucositis :Monitor at least weekly. Omit and/or reduce dose for grade 2 or higher mucositis. ( 2.4 , 5.2 ) Dermatologic reactions : Reactions, including fatal reactions, occurred and may be progressive and increase in severity with further treatment. Monitor closely and withhold or discontinue Pralatrexate injection based on severity. ( 2.4 , 5.3 ) Tumor lysis syndrome : Monitor patients who are increased risk and treat promptly. ( 5.4 ) Hepatic toxicity : Monitor for liver function tests. Omit until recovery, adjust or discontinue therapy based on severity. ( 2.4 , 5.5 ) Risk of increased toxicity with renal impairment : Avoid Pralatrexate injection in patients with end stage renal disease with or without dialysis. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions. ( 2.3 , 2.4 , 5.6 ) Embryo-fetal toxicity : Can cause fetal harm. Advise patients of the potential risk to a fetus and to use an effective method of contraception. ( 5.7 , 8.1 , 8.3 )
Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling: Myelosuppression [see Warnings and Precautions ( 5.1 )] Mucositis [see Warnings and Precautions ( 5.2 )] Dermatologic Reactions [see Warnings and Precautions ( 5.3 )] Tumor Lysis Syndrome [see Warnings and Precautions ( 5.4 )] Hepatic Toxicity [see Warnings and Precautions ( 5.5 )]
Drug Interactions
Avoid coadministration with probenecid or nonsteroidal anti-inflammatory drugs. If coadministration is unavoidable, monitor for increased risk of adverse reactions. ( 7.1 )
How Supplied
Pralatrexate Injection is available in single-dose clear glass vials containing pralatrexate at a concentration of 20 mg/mL as a preservative-free, sterile, clear yellow solution individually packaged for intravenous use in the following presentations: Product Code Unit of Sale Strength 550101 NDC 65219-550-01 20 mg/1 mL 552102 NDC 65219-552-02 40 mg/2 mL Store refrigerated at 2-8°C (36-46°F) [ see USP Controlled Cold Temperature] in original carton to protect from light. Pralatrexate Injection is a hazardous drug. Follow applicable special handling and disposal procedures. 1
Storage and Handling
Pralatrexate Injection is available in single-dose clear glass vials containing pralatrexate at a concentration of 20 mg/mL as a preservative-free, sterile, clear yellow solution individually packaged for intravenous use in the following presentations: Product Code Unit of Sale Strength 550101 NDC 65219-550-01 20 mg/1 mL 552102 NDC 65219-552-02 40 mg/2 mL Store refrigerated at 2-8°C (36-46°F) [ see USP Controlled Cold Temperature] in original carton to protect from light. Pralatrexate Injection is a hazardous drug. Follow applicable special handling and disposal procedures. 1
Description
Pralatrexate injection is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL). This indication is approved under accelerated approval based on overall response rate [see Clinical Studies ( 14 )] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Medication Information
Warnings and Precautions
Myelosuppression : Monitor complete blood counts and omit and/or reduce dose based on ANC and platelet count. ( 2.4 , 5.1 ) Mucositis :Monitor at least weekly. Omit and/or reduce dose for grade 2 or higher mucositis. ( 2.4 , 5.2 ) Dermatologic reactions : Reactions, including fatal reactions, occurred and may be progressive and increase in severity with further treatment. Monitor closely and withhold or discontinue Pralatrexate injection based on severity. ( 2.4 , 5.3 ) Tumor lysis syndrome : Monitor patients who are increased risk and treat promptly. ( 5.4 ) Hepatic toxicity : Monitor for liver function tests. Omit until recovery, adjust or discontinue therapy based on severity. ( 2.4 , 5.5 ) Risk of increased toxicity with renal impairment : Avoid Pralatrexate injection in patients with end stage renal disease with or without dialysis. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions. ( 2.3 , 2.4 , 5.6 ) Embryo-fetal toxicity : Can cause fetal harm. Advise patients of the potential risk to a fetus and to use an effective method of contraception. ( 5.7 , 8.1 , 8.3 )
Indications and Usage
Pralatrexate injection is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL). This indication is approved under accelerated approval based on overall response rate [see Clinical Studies ( 14 )] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Dosage and Administration
Supplement patients with vitamin B 12 mg intramuscularly every 8-10 weeks starting 10 weeks before the first dose and folic acid 1 to 1.25 mg orally once daily starting 10 days before the first dose. ( 2.1 ) The recommended dosage of Pralatrexate injection is 30 mg/m 2 intravenously over 3 to 5 minutes once weekly for 6 weeks in 7-week cycles. ( 2.1 ) For patients with severe renal impairment (GFR 15 to 29 mL/min/1.73 m 2 ), reduce the Pralatrexate injection dose to 15 mg/m 2 ( 2.1 ).
Contraindications
None
Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling: Myelosuppression [see Warnings and Precautions ( 5.1 )] Mucositis [see Warnings and Precautions ( 5.2 )] Dermatologic Reactions [see Warnings and Precautions ( 5.3 )] Tumor Lysis Syndrome [see Warnings and Precautions ( 5.4 )] Hepatic Toxicity [see Warnings and Precautions ( 5.5 )]
Drug Interactions
Avoid coadministration with probenecid or nonsteroidal anti-inflammatory drugs. If coadministration is unavoidable, monitor for increased risk of adverse reactions. ( 7.1 )
Storage and Handling
Pralatrexate Injection is available in single-dose clear glass vials containing pralatrexate at a concentration of 20 mg/mL as a preservative-free, sterile, clear yellow solution individually packaged for intravenous use in the following presentations: Product Code Unit of Sale Strength 550101 NDC 65219-550-01 20 mg/1 mL 552102 NDC 65219-552-02 40 mg/2 mL Store refrigerated at 2-8°C (36-46°F) [ see USP Controlled Cold Temperature] in original carton to protect from light. Pralatrexate Injection is a hazardous drug. Follow applicable special handling and disposal procedures. 1
How Supplied
Pralatrexate Injection is available in single-dose clear glass vials containing pralatrexate at a concentration of 20 mg/mL as a preservative-free, sterile, clear yellow solution individually packaged for intravenous use in the following presentations: Product Code Unit of Sale Strength 550101 NDC 65219-550-01 20 mg/1 mL 552102 NDC 65219-552-02 40 mg/2 mL Store refrigerated at 2-8°C (36-46°F) [ see USP Controlled Cold Temperature] in original carton to protect from light. Pralatrexate Injection is a hazardous drug. Follow applicable special handling and disposal procedures. 1
Description
Pralatrexate injection is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL). This indication is approved under accelerated approval based on overall response rate [see Clinical Studies ( 14 )] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Section 42229-5
Pretreatment Vitamin Supplementation
Section 42230-3
|
This Patient Information has been approved by the U.S. Food and Drug Administration. |
Revised: 09/2022 |
|
Patient Information
Pralatrexate Injection (PRA-luh-TREK-sayt) |
|
|
What is Pralatrexate injection?
Pralatrexate injection is a prescription used to treat people with a type of cancer called peripheral T-cell lymphoma (PTCL) that does not go away, gets worse, or comes back after use of another cancer treatment. It is not known if Pralatrexate injection is safe and effective in children. |
|
Before you receive Pralatrexate injection, tell your healthcare provider about all of your medical conditions, including if you:
|
|
|
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some medicines may affect how Pralatrexate injection works. Especially tell your healthcare provider if you take:
|
|
| Know the medicines you take. Keep a list of them and show it to your healthcare provider or pharmacist each time you start a new medicine. | |
How will I receive Pralatrexate injection?
|
|
| Your healthcare provider may stop treatment, delay treatment, or change your dose of Pralatrexate injection based on results of your blood tests and if you have certain side effects. | |
|
What are the possible side effects of Pralatrexate injection?
Pralatrexate injection may cause serious side effects, including:
|
|
|
The most common side effects of Pralatrexate injection include: low platelet blood counts, nausea, and tiredness. These are not all of the possible side effects of Pralatrexate injection. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
|
General information about the safe and effective use of Pralatrexate injection.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. This Patient Information leaflet summarizes the most important information about Pralatrexate injection. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about Pralatrexate injection that is written for health professionals. |
|
|
What are the ingredients in Pralatrexate injection?
Lake Zurich, IL 60047 www.fresenius-kabi.com/us Made in Germany 451757 |
Section 51945-4
PRINCIPAL DISPLAY PANEL – Pralatrexate Injection 20 mg/mL – Carton
NDC 65219-550-01
Pralatrexate
Injection
20 mg/mL
For intravenous use
Rx only
1 mL
10 Overdosage
No specific information is available on the treatment of overdosage of Pralatrexate injection. If an overdose occurs, general supportive measures should be instituted as deemed necessary by the treating healthcare provider. Based on Pralatrexate injection's mechanism of action, consider the prompt administration of leucovorin.
15 References
- “OSHA Hazardous Drugs.” OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
5.2 Mucositis
Pralatrexate injection can cause mucositis [see Adverse Reactions (6.1)].
Administer vitamin B12 and instruct patients to take folic acid to reduce the risk of mucositis [see Dosage and Administration (2.1)].
Monitor for mucositis weekly and omit and/or reduce the dose for grade 2 or higher mucositis [see Dosage and Administration (2.4)].
11 Description
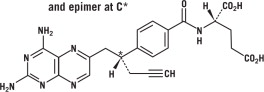
Pralatrexate is a dihydrofolate reductase inhibitor. Pralatrexate has the chemical name (2S)-2[[4-[(1RS)-1-[(2, 4-diaminopteridin-6-yl)methyl]but-3- ynyl]benzoyl]amino]pentanedioic acid. The molecular formula is C23H23N7O5 and the molecular weight is 477.48 g/mol. Pralatrexate is a 1:1 racemic mixture of S- and R- diastereomers at the C10 position (indicated with *). The structural formula is as follows:
Pralatrexate is an off-white to yellow solid. It is soluble in aqueous solutions at pH 6.5 or higher. Pralatrexate is practically insoluble in chloroform and ethanol. The pKa values are 3.25, 4.76, and 6.17.
Pralatrexate Injection is supplied as a preservative-free, sterile, isotonic, non-pyrogenic clear yellow aqueous solution contained in a clear glass single-dose vial (Type I) for intravenous use. Each 1 mL of solution contains 20 mg of pralatrexate, sufficient sodium chloride to achieve an isotonic (280-300 mOsm) solution, and sufficient sodium hydroxide, and hydrochloric acid if needed, to adjust and maintain the pH at 7.5-8.5. Pralatrexate Injection is supplied as either 20 mg (1 mL) or 40 mg (2 mL) single-dose vials at a concentration of 20 mg/mL.
8.4 Pediatric Use
The safety and effectiveness of Pralatrexate injection in pediatric patients have not been established.
8.5 Geriatric Use
In the Study PDX-008, 36% of patients (n = 40) were 65 years of age and over. No overall differences in efficacy and safety were observed in patients based on age (< 65 years compared with ≥ 65 years). Due to the contribution of renal excretion to overall clearance of pralatrexate (approximately 34%), age-related decline in renal function may lead to a reduction in clearance and a commensurate increase in plasma exposure. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. Since elderly patients may be at higher risk, monitor more closely. Omit dose and subsequently adjust or discontinue therapy for adverse reactions [see Dosage and Administration (2.4)].
14 Clinical Studies
The efficacy of Pralatrexate injection was evaluated in Study PDX-008, an open-label, single-arm, multi-center, international trial that enrolled patients with relapsed or refractory PTCL. One hundred and eleven patients received Pralatrexate injection 30 mg/m2 intravenously over 3 to 5 minutes once weekly by for 6 weeks in 7-week cycles until disease progression or unacceptable toxicity. Of the 111 patients treated, 109 patients were evaluable for efficacy. Evaluable patients had histologically confirmed PTCL by independent central review using the Revised European American Lymphoma (REAL) World Health Organization (WHO) disease classification, and relapsed or refractory disease after at least one prior treatment.
The major efficacy outcome measure was overall response rate (complete response, complete response unconfirmed, and partial response) as assessed by International Workshop Criteria (IWC). An additional efficacy outcome measure was duration of response. Response assessments were scheduled at the end of cycle 1 and then every other cycle (every 14 weeks). Duration of response was measured from the first day of documented response to disease progression or death. Response and disease progression were evaluated by independent central review using the IWC.
The median age was 59 years (range: 21 to 85); 68% were male; 72% were White, 13% were Black, 8% were Hispanic and 5% were Asian. Patients had a baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 (39%), 1 (44%), or 2 (17%). The median time from initial diagnosis to study entry was 1.3 years (range 24 days to 26.8 years). The median number of prior systemic therapies was 3 (range 1 to 12). Approximately 24% of patients (n = 27) did not have evidence of response to any previous therapy. Approximately 63% of patients (n = 70) did not have evidence of response to their most recent prior therapy before entering the study.
Efficacy results are provided in Table 5.
|
Fourteen patients went off treatment in cycle 1; 2 patients were unevaluable for response by IWC due to insufficient materials provided to central review. |
||||
|
CR = Complete Response, CRu = Complete Response unconfirmed, PR = Partial Response |
||||
|
Evaluable Patients
(N=109) |
||||
| N (%) | 95% CI | Median Duration of Response | Range of Duration of Response | |
| Overall Response | ||||
| CR+CRu+PR | 29 (27) | 19, 36 | 287 days (9.4 months) | 1-503 days |
| CR/CRu | 9 (8) | |||
| PR | 20 (18) | |||
| Responses ≥ 14 weeks | ||||
| CR+CRu+PR | 13 (12) | 7, 20 | Not Reached | 98-503 days |
| CR/CRu | 7 (6) | |||
| PR | 6 (6) |
The initial response assessment was scheduled at the end of cycle 1. Of the responders, 66% responded within cycle 1. The median time to first response was 45 days (range 37-349 days).
4 Contraindications
None
6 Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling:
7 Drug Interactions
- Avoid coadministration with probenecid or nonsteroidal anti-inflammatory drugs. If coadministration is unavoidable, monitor for increased risk of adverse reactions. (7.1)
5.1 Myelosuppression
Pralatrexate injection can cause myelosuppression, manifested by thrombocytopenia, neutropenia, and/or anemia.
Administer vitamin B12 and instruct patients to take folic acid to reduce the risk of treatment-related myelosuppression [see Dosage and Administration (2.1)].
Monitor complete blood counts and omit and/or reduce the dose based on ANC and platelet count prior to each dose [see Dosage and Administration (2.4)].
5.5 Hepatic Toxicity
Pralatrexate injection can cause hepatic toxicity and liver function test abnormalities [see Adverse Reactions (6.1)]. Persistent liver function test abnormalities may be indicators of hepatic toxicity and require dose modification or discontinuation.
Monitor liver function tests. Omit dose until recovery, adjust or discontinue therapy based on the severity of the hepatic toxicity [see Dosage and Administration (2.4)].
8.6 Renal Impairment
No dosage modification is recommended for patients with mild or moderate renal impairment (eGFR 30 to 59 mL/min/1.73 m2 based on MDRD). For patients with severe renal impairment (eGFR 15 to 29 mL/min/1.73 m2), reduce the recommended dose of Pralatrexate injection [see Dosage and Administration (2.3)].
Serious adverse drug reactions, including TEN and mucositis, have been reported in patients with ESRD undergoing dialysis. Avoid the use of Pralatrexate injection in patients with ESRD with or without dialysis. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions [see Dosage and Administration (2.3), Warnings and Precautions (5.6)].
12.2 Pharmacodynamics
Pralatrexate exposure-response relationship and the time course of pharmacodynamics responses are unknown.
12.3 Pharmacokinetics
Pralatrexate is a racemic mixture of S- and R-diastereomers. The pharmacokinetics of pralatrexate at the recommended dosage of 30 mg/m2 once weekly have been evaluated in 10 patients with PTCL. Pralatrexate total systemic exposure (AUC) and maximum plasma concentration (Cmax) increased proportionally over a dose range 30 to 325 mg/m2 (10.8 times the approved recommended dosage). No accumulation of pralatrexate was observed.
2.2 Recommended Dosage
The recommended dosage of Pralatrexate injection is 30 mg/m2 intravenously over 3-5 minutes once weekly for 6 weeks in 7-week cycles until progressive disease or unacceptable toxicity.
1 Indications and Usage
Pralatrexate injection is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
12.1 Mechanism of Action
Pralatrexate is a folate analog metabolic inhibitor that competitively inhibits dihydrofolate reductase. It is also a competitive inhibitor for polyglutamylation by the enzyme folylpolyglutamyl synthetase. This inhibition results in the depletion of thymidine and other biological molecules the synthesis of which depends on single carbon transfer.
5.4 Tumor Lysis Syndrome
Pralatrexate injection can cause tumor lysis syndrome (TLS). Monitor patients who are at increased risk of TLS and treat promptly.
5.7 Embryo Fetal Toxicity
Based on findings in animals and its mechanism of action, Pralatrexate injection can cause fetal harm when administered to a pregnant woman. Pralatrexate injection was embryotoxic and fetotoxic in rats and rabbits. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with Pralatrexate injection and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with Pralatrexate injection and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
5 Warnings and Precautions
- Myelosuppression: Monitor complete blood counts and omit and/or reduce dose based on ANC and platelet count. (2.4, 5.1)
- Mucositis:Monitor at least weekly. Omit and/or reduce dose for grade 2 or higher mucositis. (2.4, 5.2)
- Dermatologic reactions: Reactions, including fatal reactions, occurred and may be progressive and increase in severity with further treatment. Monitor closely and withhold or discontinue Pralatrexate injection based on severity. (2.4, 5.3)
- Tumor lysis syndrome: Monitor patients who are increased risk and treat promptly. (5.4)
- Hepatic toxicity: Monitor for liver function tests. Omit until recovery, adjust or discontinue therapy based on severity. (2.4, 5.5)
- Risk of increased toxicity with renal impairment: Avoid Pralatrexate injection in patients with end stage renal disease with or without dialysis. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions. (2.3, 2.4, 5.6)
- Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use an effective method of contraception. (5.7, 8.1, 8.3)
5.3 Dermatologic Reactions
Pralatrexate injection can cause severe dermatologic reactions, which may result in death. These dermatologic reactions have been reported in clinical studies (2.1% of 663 patients) and post marketing experience, and have included skin exfoliation, ulceration, and toxic epidermal necrolysis (TEN) [see Adverse Reactions (6.1, 6.2)]. They may be progressive and increase in severity with further treatment and may involve skin and subcutaneous sites of known lymphoma.
Monitor closely for dermatologic reactions. Withhold or discontinue Pralatrexate injection based on severity [see Dosage and Administration (2.4)].
2 Dosage and Administration
- Supplement patients with vitamin B12 mg intramuscularly every 8-10 weeks starting 10 weeks before the first dose and folic acid 1 to 1.25 mg orally once daily starting 10 days before the first dose. (2.1)
- The recommended dosage of Pralatrexate injection is 30 mg/m2 intravenously over 3 to 5 minutes once weekly for 6 weeks in 7-week cycles. (2.1)
- For patients with severe renal impairment (GFR 15 to 29 mL/min/1.73 m2), reduce the Pralatrexate injection dose to 15 mg/m2 (2.1).
3 Dosage Forms and Strengths
Injection: 40 mg/2 mL (20 mg/mL) and 20 mg/mL clear yellow sterile solution in single-dose vial
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Pralatrexate injection. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatologic Reactions: Toxic epidermal necrolysis.
8 Use in Specific Populations
- Lactation: Advise not to breastfeed. (8.2)
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
2.5 Preparation and Administration
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use any vials exhibiting particulate matter or discoloration.
Pralatrexate injection is a hazardous drug. Follow applicable special handling and disposal procedures.1 If Pralatrexate injection comes in contact with the skin, immediately and thoroughly wash with soap and water. If Pralatrexate injection comes in contact with mucous membranes, flush thoroughly with water.
Aseptically withdraw the calculated dose from the appropriate number of vial(s) into a syringe for immediate use. Do not dilute Pralatrexate injection.
Administer undiluted Pralatrexate injection intravenously over 3-5 minutes via the side port of a free-flowing 0.9% Sodium Chloride Injection.
After withdrawal of dose, discard vial(s) including any unused portion.
16 How Supplied/storage and Handling
Pralatrexate Injection is available in single-dose clear glass vials containing pralatrexate at a concentration of 20 mg/mL as a preservative-free, sterile, clear yellow solution individually packaged for intravenous use in the following presentations:
| Product Code | Unit of Sale | Strength |
| 550101 | NDC 65219-550-01 | 20 mg/1 mL |
| 552102 | NDC 65219-552-02 | 40 mg/2 mL |
Store refrigerated at 2-8°C (36-46°F) [see USP Controlled Cold Temperature] in original carton to protect from light.
Pralatrexate Injection is a hazardous drug. Follow applicable special handling and disposal procedures.1
8.3 Females and Males of Reproductive Potential
Pralatrexate injection can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1 )].
5.6 Risk of Increased Toxicity With Renal Impairment
Patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2 based on MDRD) may be at greater risk for increased exposure and adverse reactions. Reduce Pralatrexate injection dosage in patients with severe renal impairment [see Dosage and Administration (2.3)].
Serious adverse reactions, including TEN and mucositis, were reported in patients with end stage renal disease (ESRD) undergoing dialysis who were administered Pralatrexate injection. Avoid Pralatrexate injection in patients with ESRD with or without dialysis. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions [see Dosage and Administration (2.3)].
7.1 Effects of Other Drugs On Pralatrexate Injection
Coadministration of Pralatrexate injection with probenecid increased pralatrexate plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions. Avoid coadministration with probenecid or nonsteroidal anti-inflammatory drugs. If coadministration is unavoidable, monitor for increased risk of adverse reactions.
2.3 Dosage Modifications for Renal Impairment and End Stage Renal Disease
- Severe renal impairment (eGFR 15 to 29 mL/min/1.73 m2 by MDRD): Reduce the Pralatrexate injection dose to 15 mg/m2 [see Use in Specific Populations (8.6)].
- End stage renal disease (ESRD: eGFR less than 15 mL/min/1.73 m2 by MDRD) with or without dialysis: Avoid administration. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions [see Warnings and Precautions (5.6), Use in Specific Populations (8.6)].
Structured Label Content
Section 42229-5 (42229-5)
Pretreatment Vitamin Supplementation
Section 42230-3 (42230-3)
|
This Patient Information has been approved by the U.S. Food and Drug Administration. |
Revised: 09/2022 |
|
Patient Information
Pralatrexate Injection (PRA-luh-TREK-sayt) |
|
|
What is Pralatrexate injection?
Pralatrexate injection is a prescription used to treat people with a type of cancer called peripheral T-cell lymphoma (PTCL) that does not go away, gets worse, or comes back after use of another cancer treatment. It is not known if Pralatrexate injection is safe and effective in children. |
|
Before you receive Pralatrexate injection, tell your healthcare provider about all of your medical conditions, including if you:
|
|
|
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some medicines may affect how Pralatrexate injection works. Especially tell your healthcare provider if you take:
|
|
| Know the medicines you take. Keep a list of them and show it to your healthcare provider or pharmacist each time you start a new medicine. | |
How will I receive Pralatrexate injection?
|
|
| Your healthcare provider may stop treatment, delay treatment, or change your dose of Pralatrexate injection based on results of your blood tests and if you have certain side effects. | |
|
What are the possible side effects of Pralatrexate injection?
Pralatrexate injection may cause serious side effects, including:
|
|
|
The most common side effects of Pralatrexate injection include: low platelet blood counts, nausea, and tiredness. These are not all of the possible side effects of Pralatrexate injection. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
|
General information about the safe and effective use of Pralatrexate injection.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. This Patient Information leaflet summarizes the most important information about Pralatrexate injection. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about Pralatrexate injection that is written for health professionals. |
|
|
What are the ingredients in Pralatrexate injection?
Lake Zurich, IL 60047 www.fresenius-kabi.com/us Made in Germany 451757 |
Section 51945-4 (51945-4)
PRINCIPAL DISPLAY PANEL – Pralatrexate Injection 20 mg/mL – Carton
NDC 65219-550-01
Pralatrexate
Injection
20 mg/mL
For intravenous use
Rx only
1 mL
10 Overdosage (10 OVERDOSAGE)
No specific information is available on the treatment of overdosage of Pralatrexate injection. If an overdose occurs, general supportive measures should be instituted as deemed necessary by the treating healthcare provider. Based on Pralatrexate injection's mechanism of action, consider the prompt administration of leucovorin.
15 References (15 REFERENCES)
- “OSHA Hazardous Drugs.” OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
5.2 Mucositis
Pralatrexate injection can cause mucositis [see Adverse Reactions (6.1)].
Administer vitamin B12 and instruct patients to take folic acid to reduce the risk of mucositis [see Dosage and Administration (2.1)].
Monitor for mucositis weekly and omit and/or reduce the dose for grade 2 or higher mucositis [see Dosage and Administration (2.4)].
11 Description (11 DESCRIPTION)
Pralatrexate is a dihydrofolate reductase inhibitor. Pralatrexate has the chemical name (2S)-2[[4-[(1RS)-1-[(2, 4-diaminopteridin-6-yl)methyl]but-3- ynyl]benzoyl]amino]pentanedioic acid. The molecular formula is C23H23N7O5 and the molecular weight is 477.48 g/mol. Pralatrexate is a 1:1 racemic mixture of S- and R- diastereomers at the C10 position (indicated with *). The structural formula is as follows:
Pralatrexate is an off-white to yellow solid. It is soluble in aqueous solutions at pH 6.5 or higher. Pralatrexate is practically insoluble in chloroform and ethanol. The pKa values are 3.25, 4.76, and 6.17.
Pralatrexate Injection is supplied as a preservative-free, sterile, isotonic, non-pyrogenic clear yellow aqueous solution contained in a clear glass single-dose vial (Type I) for intravenous use. Each 1 mL of solution contains 20 mg of pralatrexate, sufficient sodium chloride to achieve an isotonic (280-300 mOsm) solution, and sufficient sodium hydroxide, and hydrochloric acid if needed, to adjust and maintain the pH at 7.5-8.5. Pralatrexate Injection is supplied as either 20 mg (1 mL) or 40 mg (2 mL) single-dose vials at a concentration of 20 mg/mL.
8.4 Pediatric Use
The safety and effectiveness of Pralatrexate injection in pediatric patients have not been established.
8.5 Geriatric Use
In the Study PDX-008, 36% of patients (n = 40) were 65 years of age and over. No overall differences in efficacy and safety were observed in patients based on age (< 65 years compared with ≥ 65 years). Due to the contribution of renal excretion to overall clearance of pralatrexate (approximately 34%), age-related decline in renal function may lead to a reduction in clearance and a commensurate increase in plasma exposure. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. Since elderly patients may be at higher risk, monitor more closely. Omit dose and subsequently adjust or discontinue therapy for adverse reactions [see Dosage and Administration (2.4)].
14 Clinical Studies (14 CLINICAL STUDIES)
The efficacy of Pralatrexate injection was evaluated in Study PDX-008, an open-label, single-arm, multi-center, international trial that enrolled patients with relapsed or refractory PTCL. One hundred and eleven patients received Pralatrexate injection 30 mg/m2 intravenously over 3 to 5 minutes once weekly by for 6 weeks in 7-week cycles until disease progression or unacceptable toxicity. Of the 111 patients treated, 109 patients were evaluable for efficacy. Evaluable patients had histologically confirmed PTCL by independent central review using the Revised European American Lymphoma (REAL) World Health Organization (WHO) disease classification, and relapsed or refractory disease after at least one prior treatment.
The major efficacy outcome measure was overall response rate (complete response, complete response unconfirmed, and partial response) as assessed by International Workshop Criteria (IWC). An additional efficacy outcome measure was duration of response. Response assessments were scheduled at the end of cycle 1 and then every other cycle (every 14 weeks). Duration of response was measured from the first day of documented response to disease progression or death. Response and disease progression were evaluated by independent central review using the IWC.
The median age was 59 years (range: 21 to 85); 68% were male; 72% were White, 13% were Black, 8% were Hispanic and 5% were Asian. Patients had a baseline Eastern Cooperative Oncology Group (ECOG) performance status of 0 (39%), 1 (44%), or 2 (17%). The median time from initial diagnosis to study entry was 1.3 years (range 24 days to 26.8 years). The median number of prior systemic therapies was 3 (range 1 to 12). Approximately 24% of patients (n = 27) did not have evidence of response to any previous therapy. Approximately 63% of patients (n = 70) did not have evidence of response to their most recent prior therapy before entering the study.
Efficacy results are provided in Table 5.
|
Fourteen patients went off treatment in cycle 1; 2 patients were unevaluable for response by IWC due to insufficient materials provided to central review. |
||||
|
CR = Complete Response, CRu = Complete Response unconfirmed, PR = Partial Response |
||||
|
Evaluable Patients
(N=109) |
||||
| N (%) | 95% CI | Median Duration of Response | Range of Duration of Response | |
| Overall Response | ||||
| CR+CRu+PR | 29 (27) | 19, 36 | 287 days (9.4 months) | 1-503 days |
| CR/CRu | 9 (8) | |||
| PR | 20 (18) | |||
| Responses ≥ 14 weeks | ||||
| CR+CRu+PR | 13 (12) | 7, 20 | Not Reached | 98-503 days |
| CR/CRu | 7 (6) | |||
| PR | 6 (6) |
The initial response assessment was scheduled at the end of cycle 1. Of the responders, 66% responded within cycle 1. The median time to first response was 45 days (range 37-349 days).
4 Contraindications (4 CONTRAINDICATIONS)
None
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following clinically significant adverse reactions are described elsewhere in the labeling:
7 Drug Interactions (7 DRUG INTERACTIONS)
- Avoid coadministration with probenecid or nonsteroidal anti-inflammatory drugs. If coadministration is unavoidable, monitor for increased risk of adverse reactions. (7.1)
5.1 Myelosuppression
Pralatrexate injection can cause myelosuppression, manifested by thrombocytopenia, neutropenia, and/or anemia.
Administer vitamin B12 and instruct patients to take folic acid to reduce the risk of treatment-related myelosuppression [see Dosage and Administration (2.1)].
Monitor complete blood counts and omit and/or reduce the dose based on ANC and platelet count prior to each dose [see Dosage and Administration (2.4)].
5.5 Hepatic Toxicity
Pralatrexate injection can cause hepatic toxicity and liver function test abnormalities [see Adverse Reactions (6.1)]. Persistent liver function test abnormalities may be indicators of hepatic toxicity and require dose modification or discontinuation.
Monitor liver function tests. Omit dose until recovery, adjust or discontinue therapy based on the severity of the hepatic toxicity [see Dosage and Administration (2.4)].
8.6 Renal Impairment
No dosage modification is recommended for patients with mild or moderate renal impairment (eGFR 30 to 59 mL/min/1.73 m2 based on MDRD). For patients with severe renal impairment (eGFR 15 to 29 mL/min/1.73 m2), reduce the recommended dose of Pralatrexate injection [see Dosage and Administration (2.3)].
Serious adverse drug reactions, including TEN and mucositis, have been reported in patients with ESRD undergoing dialysis. Avoid the use of Pralatrexate injection in patients with ESRD with or without dialysis. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions [see Dosage and Administration (2.3), Warnings and Precautions (5.6)].
12.2 Pharmacodynamics
Pralatrexate exposure-response relationship and the time course of pharmacodynamics responses are unknown.
12.3 Pharmacokinetics
Pralatrexate is a racemic mixture of S- and R-diastereomers. The pharmacokinetics of pralatrexate at the recommended dosage of 30 mg/m2 once weekly have been evaluated in 10 patients with PTCL. Pralatrexate total systemic exposure (AUC) and maximum plasma concentration (Cmax) increased proportionally over a dose range 30 to 325 mg/m2 (10.8 times the approved recommended dosage). No accumulation of pralatrexate was observed.
2.2 Recommended Dosage
The recommended dosage of Pralatrexate injection is 30 mg/m2 intravenously over 3-5 minutes once weekly for 6 weeks in 7-week cycles until progressive disease or unacceptable toxicity.
1 Indications and Usage (1 INDICATIONS AND USAGE)
Pralatrexate injection is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL).
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
12.1 Mechanism of Action
Pralatrexate is a folate analog metabolic inhibitor that competitively inhibits dihydrofolate reductase. It is also a competitive inhibitor for polyglutamylation by the enzyme folylpolyglutamyl synthetase. This inhibition results in the depletion of thymidine and other biological molecules the synthesis of which depends on single carbon transfer.
5.4 Tumor Lysis Syndrome
Pralatrexate injection can cause tumor lysis syndrome (TLS). Monitor patients who are at increased risk of TLS and treat promptly.
5.7 Embryo Fetal Toxicity (5.7 Embryo-Fetal Toxicity)
Based on findings in animals and its mechanism of action, Pralatrexate injection can cause fetal harm when administered to a pregnant woman. Pralatrexate injection was embryotoxic and fetotoxic in rats and rabbits. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with Pralatrexate injection and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with Pralatrexate injection and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Myelosuppression: Monitor complete blood counts and omit and/or reduce dose based on ANC and platelet count. (2.4, 5.1)
- Mucositis:Monitor at least weekly. Omit and/or reduce dose for grade 2 or higher mucositis. (2.4, 5.2)
- Dermatologic reactions: Reactions, including fatal reactions, occurred and may be progressive and increase in severity with further treatment. Monitor closely and withhold or discontinue Pralatrexate injection based on severity. (2.4, 5.3)
- Tumor lysis syndrome: Monitor patients who are increased risk and treat promptly. (5.4)
- Hepatic toxicity: Monitor for liver function tests. Omit until recovery, adjust or discontinue therapy based on severity. (2.4, 5.5)
- Risk of increased toxicity with renal impairment: Avoid Pralatrexate injection in patients with end stage renal disease with or without dialysis. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions. (2.3, 2.4, 5.6)
- Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use an effective method of contraception. (5.7, 8.1, 8.3)
5.3 Dermatologic Reactions
Pralatrexate injection can cause severe dermatologic reactions, which may result in death. These dermatologic reactions have been reported in clinical studies (2.1% of 663 patients) and post marketing experience, and have included skin exfoliation, ulceration, and toxic epidermal necrolysis (TEN) [see Adverse Reactions (6.1, 6.2)]. They may be progressive and increase in severity with further treatment and may involve skin and subcutaneous sites of known lymphoma.
Monitor closely for dermatologic reactions. Withhold or discontinue Pralatrexate injection based on severity [see Dosage and Administration (2.4)].
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
- Supplement patients with vitamin B12 mg intramuscularly every 8-10 weeks starting 10 weeks before the first dose and folic acid 1 to 1.25 mg orally once daily starting 10 days before the first dose. (2.1)
- The recommended dosage of Pralatrexate injection is 30 mg/m2 intravenously over 3 to 5 minutes once weekly for 6 weeks in 7-week cycles. (2.1)
- For patients with severe renal impairment (GFR 15 to 29 mL/min/1.73 m2), reduce the Pralatrexate injection dose to 15 mg/m2 (2.1).
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Injection: 40 mg/2 mL (20 mg/mL) and 20 mg/mL clear yellow sterile solution in single-dose vial
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Pralatrexate injection. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatologic Reactions: Toxic epidermal necrolysis.
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
- Lactation: Advise not to breastfeed. (8.2)
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA-approved patient labeling (Patient Information).
2.5 Preparation and Administration
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use any vials exhibiting particulate matter or discoloration.
Pralatrexate injection is a hazardous drug. Follow applicable special handling and disposal procedures.1 If Pralatrexate injection comes in contact with the skin, immediately and thoroughly wash with soap and water. If Pralatrexate injection comes in contact with mucous membranes, flush thoroughly with water.
Aseptically withdraw the calculated dose from the appropriate number of vial(s) into a syringe for immediate use. Do not dilute Pralatrexate injection.
Administer undiluted Pralatrexate injection intravenously over 3-5 minutes via the side port of a free-flowing 0.9% Sodium Chloride Injection.
After withdrawal of dose, discard vial(s) including any unused portion.
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
Pralatrexate Injection is available in single-dose clear glass vials containing pralatrexate at a concentration of 20 mg/mL as a preservative-free, sterile, clear yellow solution individually packaged for intravenous use in the following presentations:
| Product Code | Unit of Sale | Strength |
| 550101 | NDC 65219-550-01 | 20 mg/1 mL |
| 552102 | NDC 65219-552-02 | 40 mg/2 mL |
Store refrigerated at 2-8°C (36-46°F) [see USP Controlled Cold Temperature] in original carton to protect from light.
Pralatrexate Injection is a hazardous drug. Follow applicable special handling and disposal procedures.1
8.3 Females and Males of Reproductive Potential
Pralatrexate injection can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1 )].
5.6 Risk of Increased Toxicity With Renal Impairment (5.6 Risk of Increased Toxicity with Renal Impairment)
Patients with severe renal impairment (eGFR 15 to < 30 mL/min/1.73 m2 based on MDRD) may be at greater risk for increased exposure and adverse reactions. Reduce Pralatrexate injection dosage in patients with severe renal impairment [see Dosage and Administration (2.3)].
Serious adverse reactions, including TEN and mucositis, were reported in patients with end stage renal disease (ESRD) undergoing dialysis who were administered Pralatrexate injection. Avoid Pralatrexate injection in patients with ESRD with or without dialysis. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions [see Dosage and Administration (2.3)].
7.1 Effects of Other Drugs On Pralatrexate Injection (7.1 Effects of Other Drugs on Pralatrexate Injection)
Coadministration of Pralatrexate injection with probenecid increased pralatrexate plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions. Avoid coadministration with probenecid or nonsteroidal anti-inflammatory drugs. If coadministration is unavoidable, monitor for increased risk of adverse reactions.
2.3 Dosage Modifications for Renal Impairment and End Stage Renal Disease
- Severe renal impairment (eGFR 15 to 29 mL/min/1.73 m2 by MDRD): Reduce the Pralatrexate injection dose to 15 mg/m2 [see Use in Specific Populations (8.6)].
- End stage renal disease (ESRD: eGFR less than 15 mL/min/1.73 m2 by MDRD) with or without dialysis: Avoid administration. If the potential benefit of administration justifies the potential risk, monitor renal function and reduce the Pralatrexate injection dose based on adverse reactions [see Warnings and Precautions (5.6), Use in Specific Populations (8.6)].
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:37:28.883764 · Updated: 2026-03-14T21:50:25.596456