Sutent
a5d555f5-d66a-4f94-abcf-96fa6d71a32f
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
SUTENT is a kinase inhibitor indicated for: • treatment of adult patients with gastrointestinal stromal tumor (GIST) after disease progression on or intolerance to imatinib mesylate. ( 1.1 ) • treatment of adult patients with advanced renal cell carcinoma (RCC). ( 1.2 ) • adjuvant treatment of adult patients at high risk of recurrent RCC following nephrectomy. ( 1.3 ) • treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNET) in adult patients with unresectable locally advanced or metastatic disease. ( 1.4 )
Dosage and Administration
GIST and Advanced RCC : • The recommended dosage is 50 mg orally once daily for the first 4 weeks of each 6-week cycle (Schedule 4/2). ( 2.1 ) Adjuvant Treatment of RCC : • The recommended dosage is 50 mg orally once daily for the first 4 weeks of a 6-week cycle (Schedule 4/2) for a maximum of 9 cycles. ( 2.2 ) pNET : • The recommended dosage is 37.5 mg orally once daily. ( 2.3 )
Contraindications
None.
Warnings and Precautions
• Hepatotoxicity : Fatal liver failure has been observed. Monitor liver function tests at baseline, during each cycle, and as clinically indicated. Interrupt SUTENT for Grade 3 hepatotoxicity until resolution to Grade ≤1 or baseline and resume SUTENT at a reduced dose; discontinue if no resolution. Discontinue SUTENT in patients with Grade 4 hepatoxicity, in patients who have subsequent severe changes in liver function tests or other signs and symptoms of liver failure. ( 2.4 , 5.1 ) • Cardiovascular Events : Myocardial ischemia, myocardial infarction, heart failure, cardiomyopathy, and decreased left ventricular ejection fraction (LVEF) to below the lower limit of normal including death have occurred. Monitor for signs and symptoms of congestive heart failure and consider monitoring LVEF at baseline and periodically during treatment. Discontinue SUTENT for clinical manifestations of congestive heart failure. Interrupt and/or dose reduce for decreased LVEF. ( 5.2 ) • QT Interval Prolongation and Torsade de Pointes : Monitor patients at higher risk for developing QT interval prolongation. Consider monitoring of electrocardiograms and electrolytes. ( 5.3 ) • Hypertension : Monitor blood pressure at baseline and as clinically indicated. Initiate and/or adjust antihypertensive therapy as appropriate. Interrupt SUTENT for Grade 3 hypertension until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose. Discontinue SUTENT in patients who develop Grade 4 hypertension. ( 5.4 ) • Hemorrhagic Events : Tumor-related hemorrhage and viscus perforation (both with fatal events) have occurred. Perform serial complete blood counts and physical examinations. Interrupt SUTENT for Grade 3 or 4 hemorrhagic events until resolution to Grade ≤1 or baseline, then resume at a reduced dose; discontinue if no resolution. ( 5.5 ) • Tumor Lysis Syndrome (TLS) : TLS (some fatal) has been reported primarily in patients with RCC and GIST. Monitor these patients and treat as clinically indicated. ( 5.6 ) • Thrombotic microangiopathy (TMA) : TMA, including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome, sometimes leading to renal failure or a fatal outcome, has been reported. Discontinue SUTENT for TMA. ( 5.7 ) • Proteinuria : Renal failure or a fatal outcome has occurred. Monitor urine protein. Interrupt treatment for 24-hour urine protein of 3 or more grams. Discontinue for repeat episodes of 24-hour urine protein of 3 or more grams despite dose reductions or nephrotic syndrome. ( 5.8 ) • Dermatologic Toxicities : Necrotizing fasciitis, erythema multiforme, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) (some fatal) have occurred. Discontinue SUTENT for these events. ( 5.9 ) • Reversible Posterior Leukoencephalopathy Syndrome (RPLS) : RPLS (some fatal) has been reported. Monitor for signs and symptoms of RPLS. Withhold SUTENT until resolution. ( 5.10 ) • Thyroid Dysfunction : Monitor thyroid function at baseline, periodically during treatment, and as clinically indicated. Initiate and/or adjust therapy for thyroid dysfunction as appropriate. ( 5.11 ) • Hypoglycemia : Check blood glucose levels regularly and assess if antidiabetic drug dose modifications are required. ( 5.12 ) • Osteonecrosis of the Jaw (ONJ) : Withhold SUTENT for at least 3 weeks prior to invasive dental procedure and for development of ONJ until complete resolution. ( 5.13 ) • Impaired Wound Healing : Withhold SUTENT for at least 3 weeks prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of SUTENT after resolution of wound healing complications has not been established. ( 5.14 ) • Embryo-Fetal Toxicity : Can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. ( 5.15 , 8.1 , 8.3 )
Adverse Reactions
To manage adverse reactions, the recommended dosage modifications are provided in Table 1. Table 2 provides the recommended dosage reductions of SUTENT for adverse reactions. Table 1. Recommended Dosage Reductions of SUTENT for Adverse Reactions Indications GIST RCC pNET Advanced RCC Adjuvant RCC First dose reduction 37.5 mg once daily 37.5 mg once daily 37.5 mg once daily 25 mg once daily Second dose reduction 25 mg once daily 25 mg once daily NA NA Table 2. Recommended Dosage Modifications for SUTENT for Adverse Reactions Adverse Reaction Severity Dosage Modifications for SUTENT Hepatotoxicity [see Warnings and Precautions (5.1) ] Grade 3 • Withhold until resolution to Grade 0 to 1 or baseline. • Resume at a reduced dose. • For recurring Grade 3 permanently discontinue. Grade 4 • Permanently discontinue. Cardiovascular events [see Warnings and Precautions (5.2) ] Asymptomatic cardiomyopathy (left ventricular ejection fraction greater than 20% but less than 50% below baseline or below the lower limit of normal if baseline was not obtained) • Withhold until resolution to Grade 0 to 1 or baseline. • Resume at reduced dose. Clinically manifested congestive heart failure (CHF) • Permanently discontinue. Hypertension [see Warnings and Precautions (5.4) ] Grade 3 • Withhold until resolution to Grade 0 to 1 or baseline. • Resume at a reduced dose. Grade 4 • Permanently discontinue. Hemorrhagic events [see Warnings and Precautions (5.5) ] Grade 3 or 4 • Withhold until resolution to Grade 0 to 1 or baseline. • Either resume at a reduced dose or discontinue depending on the severity and persistence of adverse reaction. Thrombotic microangiopathy [see Warnings and Precautions (5.7) ] Any Grade • Permanently discontinue. Proteinuria or Nephrotic syndrome [see Warnings and Precautions (5.8) ] 3 or more grams proteinuria in 24 hours in the absence of nephrotic syndrome • Withhold until resolution to Grade 0 to 1 or baseline. • Resume at a reduced dose. Nephrotic syndrome or recurrent proteinuria of 3 or more grams per 24 hours despite dose reductions • Permanently discontinue. Dermatological toxicities Erythema multiforme (EM), Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), Necrotizing fasciitis [see Warnings and Precautions (5.9) ] Any Grade • Permanently discontinue. Reversible posterior leukoencephalopathy syndrome [see Warnings and Precautions (5.10) ] Any Grade • Permanently discontinue. Osteonecrosis of the jaw [see Warnings and Precautions (5.13) ] Any Grade • The safety of resumption of SUTENT after osteonecrosis has not been established. • Either resume at a reduced dose or discontinue depending on the severity and persistence of the adverse reaction. Impaired wound healing [see Warnings and Precautions (5.14) ] Any Grade • The safety of resumption of SUTENT after resolution of wound healing has not been established. • Either resume at a reduced dose or discontinue depending on the severity and persistence of the adverse reaction.
Drug Interactions
• CYP3A4 Inhibitors : Consider dose reduction of SUTENT when administered with strong CYP3A4 inhibitors. ( 7.1 ) • CYP3A4 Inducers : Consider dose increase of SUTENT when administered with strong CYP3A4 inducers. ( 7.1 )
How Supplied
SUTENT 12.5 mg capsules are supplied as hard gelatin capsule with orange cap and orange body, printed with white ink "Pfizer" on the cap, "STN 12.5 mg" on the body: Bottles of 28 capsules: NDC 0069-0550-38 SUTENT 25 mg capsules are supplied as hard gelatin capsule with caramel cap and orange body, printed with white ink "Pfizer" on the cap, "STN 25 mg" on the body: Bottles of 28 capsules: NDC 0069-0770-38 SUTENT 37.5 mg capsules are supplied as hard gelatin capsule with yellow cap and yellow body, printed with black ink "Pfizer" on the cap, "STN 37.5 mg" on the body: Bottles of 28 capsules: NDC 0069-0830-38 SUTENT 50 mg capsules are supplied as hard gelatin capsule with caramel cap and caramel body, printed with white ink "Pfizer" on the cap, "STN 50 mg" on the body: Bottles of 28 capsules: NDC 0069-0980-38
Storage and Handling
SUTENT 12.5 mg capsules are supplied as hard gelatin capsule with orange cap and orange body, printed with white ink "Pfizer" on the cap, "STN 12.5 mg" on the body: Bottles of 28 capsules: NDC 0069-0550-38 SUTENT 25 mg capsules are supplied as hard gelatin capsule with caramel cap and orange body, printed with white ink "Pfizer" on the cap, "STN 25 mg" on the body: Bottles of 28 capsules: NDC 0069-0770-38 SUTENT 37.5 mg capsules are supplied as hard gelatin capsule with yellow cap and yellow body, printed with black ink "Pfizer" on the cap, "STN 37.5 mg" on the body: Bottles of 28 capsules: NDC 0069-0830-38 SUTENT 50 mg capsules are supplied as hard gelatin capsule with caramel cap and caramel body, printed with white ink "Pfizer" on the cap, "STN 50 mg" on the body: Bottles of 28 capsules: NDC 0069-0980-38
Description
Hepatotoxicity may be severe, and in some cases, fatal. Monitor hepatic function and interrupt, dose reduce, or discontinue SUTENT as recommended [see Warnings and Precautions (5.1) ] .
Medication Information
Warnings and Precautions
• Hepatotoxicity : Fatal liver failure has been observed. Monitor liver function tests at baseline, during each cycle, and as clinically indicated. Interrupt SUTENT for Grade 3 hepatotoxicity until resolution to Grade ≤1 or baseline and resume SUTENT at a reduced dose; discontinue if no resolution. Discontinue SUTENT in patients with Grade 4 hepatoxicity, in patients who have subsequent severe changes in liver function tests or other signs and symptoms of liver failure. ( 2.4 , 5.1 ) • Cardiovascular Events : Myocardial ischemia, myocardial infarction, heart failure, cardiomyopathy, and decreased left ventricular ejection fraction (LVEF) to below the lower limit of normal including death have occurred. Monitor for signs and symptoms of congestive heart failure and consider monitoring LVEF at baseline and periodically during treatment. Discontinue SUTENT for clinical manifestations of congestive heart failure. Interrupt and/or dose reduce for decreased LVEF. ( 5.2 ) • QT Interval Prolongation and Torsade de Pointes : Monitor patients at higher risk for developing QT interval prolongation. Consider monitoring of electrocardiograms and electrolytes. ( 5.3 ) • Hypertension : Monitor blood pressure at baseline and as clinically indicated. Initiate and/or adjust antihypertensive therapy as appropriate. Interrupt SUTENT for Grade 3 hypertension until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose. Discontinue SUTENT in patients who develop Grade 4 hypertension. ( 5.4 ) • Hemorrhagic Events : Tumor-related hemorrhage and viscus perforation (both with fatal events) have occurred. Perform serial complete blood counts and physical examinations. Interrupt SUTENT for Grade 3 or 4 hemorrhagic events until resolution to Grade ≤1 or baseline, then resume at a reduced dose; discontinue if no resolution. ( 5.5 ) • Tumor Lysis Syndrome (TLS) : TLS (some fatal) has been reported primarily in patients with RCC and GIST. Monitor these patients and treat as clinically indicated. ( 5.6 ) • Thrombotic microangiopathy (TMA) : TMA, including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome, sometimes leading to renal failure or a fatal outcome, has been reported. Discontinue SUTENT for TMA. ( 5.7 ) • Proteinuria : Renal failure or a fatal outcome has occurred. Monitor urine protein. Interrupt treatment for 24-hour urine protein of 3 or more grams. Discontinue for repeat episodes of 24-hour urine protein of 3 or more grams despite dose reductions or nephrotic syndrome. ( 5.8 ) • Dermatologic Toxicities : Necrotizing fasciitis, erythema multiforme, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) (some fatal) have occurred. Discontinue SUTENT for these events. ( 5.9 ) • Reversible Posterior Leukoencephalopathy Syndrome (RPLS) : RPLS (some fatal) has been reported. Monitor for signs and symptoms of RPLS. Withhold SUTENT until resolution. ( 5.10 ) • Thyroid Dysfunction : Monitor thyroid function at baseline, periodically during treatment, and as clinically indicated. Initiate and/or adjust therapy for thyroid dysfunction as appropriate. ( 5.11 ) • Hypoglycemia : Check blood glucose levels regularly and assess if antidiabetic drug dose modifications are required. ( 5.12 ) • Osteonecrosis of the Jaw (ONJ) : Withhold SUTENT for at least 3 weeks prior to invasive dental procedure and for development of ONJ until complete resolution. ( 5.13 ) • Impaired Wound Healing : Withhold SUTENT for at least 3 weeks prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of SUTENT after resolution of wound healing complications has not been established. ( 5.14 ) • Embryo-Fetal Toxicity : Can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. ( 5.15 , 8.1 , 8.3 )
Indications and Usage
SUTENT is a kinase inhibitor indicated for: • treatment of adult patients with gastrointestinal stromal tumor (GIST) after disease progression on or intolerance to imatinib mesylate. ( 1.1 ) • treatment of adult patients with advanced renal cell carcinoma (RCC). ( 1.2 ) • adjuvant treatment of adult patients at high risk of recurrent RCC following nephrectomy. ( 1.3 ) • treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNET) in adult patients with unresectable locally advanced or metastatic disease. ( 1.4 )
Dosage and Administration
GIST and Advanced RCC : • The recommended dosage is 50 mg orally once daily for the first 4 weeks of each 6-week cycle (Schedule 4/2). ( 2.1 ) Adjuvant Treatment of RCC : • The recommended dosage is 50 mg orally once daily for the first 4 weeks of a 6-week cycle (Schedule 4/2) for a maximum of 9 cycles. ( 2.2 ) pNET : • The recommended dosage is 37.5 mg orally once daily. ( 2.3 )
Contraindications
None.
Adverse Reactions
To manage adverse reactions, the recommended dosage modifications are provided in Table 1. Table 2 provides the recommended dosage reductions of SUTENT for adverse reactions. Table 1. Recommended Dosage Reductions of SUTENT for Adverse Reactions Indications GIST RCC pNET Advanced RCC Adjuvant RCC First dose reduction 37.5 mg once daily 37.5 mg once daily 37.5 mg once daily 25 mg once daily Second dose reduction 25 mg once daily 25 mg once daily NA NA Table 2. Recommended Dosage Modifications for SUTENT for Adverse Reactions Adverse Reaction Severity Dosage Modifications for SUTENT Hepatotoxicity [see Warnings and Precautions (5.1) ] Grade 3 • Withhold until resolution to Grade 0 to 1 or baseline. • Resume at a reduced dose. • For recurring Grade 3 permanently discontinue. Grade 4 • Permanently discontinue. Cardiovascular events [see Warnings and Precautions (5.2) ] Asymptomatic cardiomyopathy (left ventricular ejection fraction greater than 20% but less than 50% below baseline or below the lower limit of normal if baseline was not obtained) • Withhold until resolution to Grade 0 to 1 or baseline. • Resume at reduced dose. Clinically manifested congestive heart failure (CHF) • Permanently discontinue. Hypertension [see Warnings and Precautions (5.4) ] Grade 3 • Withhold until resolution to Grade 0 to 1 or baseline. • Resume at a reduced dose. Grade 4 • Permanently discontinue. Hemorrhagic events [see Warnings and Precautions (5.5) ] Grade 3 or 4 • Withhold until resolution to Grade 0 to 1 or baseline. • Either resume at a reduced dose or discontinue depending on the severity and persistence of adverse reaction. Thrombotic microangiopathy [see Warnings and Precautions (5.7) ] Any Grade • Permanently discontinue. Proteinuria or Nephrotic syndrome [see Warnings and Precautions (5.8) ] 3 or more grams proteinuria in 24 hours in the absence of nephrotic syndrome • Withhold until resolution to Grade 0 to 1 or baseline. • Resume at a reduced dose. Nephrotic syndrome or recurrent proteinuria of 3 or more grams per 24 hours despite dose reductions • Permanently discontinue. Dermatological toxicities Erythema multiforme (EM), Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), Necrotizing fasciitis [see Warnings and Precautions (5.9) ] Any Grade • Permanently discontinue. Reversible posterior leukoencephalopathy syndrome [see Warnings and Precautions (5.10) ] Any Grade • Permanently discontinue. Osteonecrosis of the jaw [see Warnings and Precautions (5.13) ] Any Grade • The safety of resumption of SUTENT after osteonecrosis has not been established. • Either resume at a reduced dose or discontinue depending on the severity and persistence of the adverse reaction. Impaired wound healing [see Warnings and Precautions (5.14) ] Any Grade • The safety of resumption of SUTENT after resolution of wound healing has not been established. • Either resume at a reduced dose or discontinue depending on the severity and persistence of the adverse reaction.
Drug Interactions
• CYP3A4 Inhibitors : Consider dose reduction of SUTENT when administered with strong CYP3A4 inhibitors. ( 7.1 ) • CYP3A4 Inducers : Consider dose increase of SUTENT when administered with strong CYP3A4 inducers. ( 7.1 )
Storage and Handling
SUTENT 12.5 mg capsules are supplied as hard gelatin capsule with orange cap and orange body, printed with white ink "Pfizer" on the cap, "STN 12.5 mg" on the body: Bottles of 28 capsules: NDC 0069-0550-38 SUTENT 25 mg capsules are supplied as hard gelatin capsule with caramel cap and orange body, printed with white ink "Pfizer" on the cap, "STN 25 mg" on the body: Bottles of 28 capsules: NDC 0069-0770-38 SUTENT 37.5 mg capsules are supplied as hard gelatin capsule with yellow cap and yellow body, printed with black ink "Pfizer" on the cap, "STN 37.5 mg" on the body: Bottles of 28 capsules: NDC 0069-0830-38 SUTENT 50 mg capsules are supplied as hard gelatin capsule with caramel cap and caramel body, printed with white ink "Pfizer" on the cap, "STN 50 mg" on the body: Bottles of 28 capsules: NDC 0069-0980-38
How Supplied
SUTENT 12.5 mg capsules are supplied as hard gelatin capsule with orange cap and orange body, printed with white ink "Pfizer" on the cap, "STN 12.5 mg" on the body: Bottles of 28 capsules: NDC 0069-0550-38 SUTENT 25 mg capsules are supplied as hard gelatin capsule with caramel cap and orange body, printed with white ink "Pfizer" on the cap, "STN 25 mg" on the body: Bottles of 28 capsules: NDC 0069-0770-38 SUTENT 37.5 mg capsules are supplied as hard gelatin capsule with yellow cap and yellow body, printed with black ink "Pfizer" on the cap, "STN 37.5 mg" on the body: Bottles of 28 capsules: NDC 0069-0830-38 SUTENT 50 mg capsules are supplied as hard gelatin capsule with caramel cap and caramel body, printed with white ink "Pfizer" on the cap, "STN 50 mg" on the body: Bottles of 28 capsules: NDC 0069-0980-38
Description
Hepatotoxicity may be severe, and in some cases, fatal. Monitor hepatic function and interrupt, dose reduce, or discontinue SUTENT as recommended [see Warnings and Precautions (5.1) ] .
Section 42229-5
Strong CYP3A4 Inhibitors
Select an alternate concomitant medication with no or minimal enzyme inhibition potential. If coadministration of SUTENT with a strong CYP3A4 inhibitor cannot be avoided, consider a dose reduction for SUTENT to a minimum dosage as follows [see Drug Interactions (7.1)]:
-
•GIST and RCC: 37.5 mg orally once daily, on a schedule of 4 weeks on treatment followed by 2 weeks off (Schedule 4/2)
-
•pNET: 25 mg orally once daily
Section 42231-1
| This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: October 2025 | ||
|
MEDICATION GUIDE
|
||
|
What is the most important information I should know about SUTENT?
Your healthcare provider should do blood tests to check your liver function before you start taking and during treatment with SUTENT. Your healthcare provider may temporarily stop, reduce your dose, or permanently stop treatment with SUTENT if you develop liver problems. |
||
|
What is SUTENT?
It is not known if SUTENT is safe and effective in children. |
||
|
Before taking SUTENT tell your healthcare provider about all of your medical conditions, including if you:
Tell all of your healthcare providers and dentists that you are taking SUTENT. They should talk to the healthcare provider who prescribed SUTENT for you, before you have any surgery, or medical or dental procedure. |
||
|
How should I take SUTENT?
|
||
|
What are possible side effects of SUTENT?
|
||
|
|
|
|
||
|
|
|
|
Your healthcare provider may prescribe medicine for you to treat high blood pressure, if needed. |
||
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
Your healthcare provider may temporarily stop, reduce your dose, or permanently stop treatment with SUTENT if you develop serious side effects.
|
||
|
|
|
|
The medicine in SUTENT is yellow, and it may make your skin look yellow. Your skin and hair may get lighter in color. SUTENT may also cause other skin problems including: dryness, thickness or cracking of the skin. |
||
|
How do I store SUTENT?
Keep SUTENT and all medicines out of the reach of children. |
||
|
General information about the safe and effective use of SUTENT.
|
||
|
What are the ingredients in SUTENT?
LAB-0361-13.0 |
Section 44425-7
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
10 Overdosage
Treatment of overdose with SUTENT should consist of general supportive measures. There is no specific antidote for overdosage with SUTENT. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage. Cases of accidental overdose have been reported; these cases were associated with adverse reactions consistent with the known safety profile of SUTENT, or without adverse reactions. In nonclinical studies, mortality was observed following as few as 5 daily doses of 500 mg/kg (3000 mg/m2) in rats. At this dose, signs of toxicity included impaired muscle coordination, head shakes, hypoactivity, ocular discharge, piloerection, and gastrointestinal distress. Mortality and similar signs of toxicity were observed at lower doses when administered for longer durations.
8.2 Lactation
There is no information regarding the presence of sunitinib and its metabolites in human milk. Sunitinib and its metabolites were excreted in rat milk at concentrations up to 12-fold higher than in plasma (see Data). Because of the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with SUTENT and for at least 4 weeks after the last dose.
11 Description
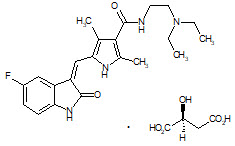
Sunitinib is a kinase inhibitor present in SUTENT capsules as the malate salt. Sunitinib malate is described chemically as (2S)-2-hydroxybutanedoic acid with N-[2-(diethylamino)ethyl]-5-[(Z)-(5-fluoro-1,2-dihydro-2-oxo-3H-indol-3-ylidine)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide (1:1). The molecular formula is C22H27FN4O2 ∙ C4H6O5 and the molecular weight is 532.6 Daltons. The chemical structure of sunitinib malate is:
Sunitinib malate is a yellow to orange powder with a pKa of 8.95. The solubility of sunitinib malate in aqueous media over the range pH 1.2 to pH 6.8 is in excess of 25 mg/mL. The log of the distribution coefficient (octanol/water) at pH 7 is 5.2.
SUTENT (sunitinib malate) capsules are supplied as printed hard shell capsules containing 12.5 mg, 25 mg, 37.5 mg or 50 mg of sunitinib (equivalent to 16.7 mg, 33.4 mg, 50.1 mg, or 66.8 mg of sunitinib malate, respectively). The capsules contain the following inactive ingredients: croscarmellose sodium, magnesium stearate, mannitol, and povidone (K-25). The orange gelatin capsule shells contain titanium dioxide and red iron oxide; the caramel gelatin capsule shells contain titanium dioxide, red iron oxide, yellow iron oxide, and black iron oxide; and the yellow gelatin capsule shells contain titanium dioxide and yellow iron oxide. The white printing ink contains shellac, propylene glycol, sodium hydroxide, povidone, and titanium dioxide and the black printing ink contains shellac, propylene glycol, potassium hydroxide, and black iron oxide.
5.8 Proteinuria
Proteinuria and nephrotic syndrome have been reported. Some of these cases have resulted in renal failure and fatal outcomes.
Monitor patients for the development or worsening of proteinuria. Perform baseline and periodic urinalyses during treatment, with follow up measurement of 24-hour urine protein as clinically indicated. Interrupt SUTENT and dose reduce for 24-hour urine protein of 3 or more grams. Discontinue SUTENT for patients with nephrotic syndrome or repeat episodes of 24-hour urine protein of 3 or more grams despite dose reductions. The safety of continued SUTENT treatment in patients with moderate to severe proteinuria has not been evaluated.
5.4 Hypertension
In the pooled safety population, 29% of patients experienced hypertension. Grade 3 hypertension was reported in 7% of patients, and Grade 4 hypertension was reported in 0.2%.
Monitor blood pressure at baseline and as clinically indicated. Initiate and/or adjust antihypertensive therapy as appropriate. In cases of Grade 3 hypertension, withhold SUTENT until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose. Discontinue SUTENT in patients with who develop Grade 4 hypertension.
5.12 Hypoglycemia
SUTENT can result in symptomatic hypoglycemia, which may lead to loss of consciousness, or require hospitalization. In the pooled safety population, hypoglycemia occurred in 2% of the patients treated with SUTENT. Hypoglycemia has occurred in clinical trials in 2% of the patients treated with SUTENT for advanced RCC (Study 3) and GIST (Study 1) (n=577) and in approximately 10% of the patients treated with SUTENT for pNET (Study 6) (n=83). For patients being treated with SUTENT for pNET, pre-existing abnormalities in glucose homeostasis were not present in all patients who experienced hypoglycemia. Reductions in blood glucose levels may be worse in patients with diabetes.
Check blood glucose levels at baseline, regularly during treatment, as clinically indicated and after discontinuation of SUTENT. In patients with diabetes, assess if antidiabetic therapies need to be adjusted to minimize the risk of hypoglycemia.
8.4 Pediatric Use
The safety and effectiveness of SUTENT in pediatric patients have not been established. Safety and pharmacokinetics of sunitinib were assessed in an open-label study (NCT00387920) in pediatric patients 2 years to <17 years of age (n=29) with refractory solid tumors. In addition, efficacy, safety and pharmacokinetics of sunitinib was assessed in another open-label study (NCT01462695) in pediatric patients 2 years to <17 years of age (n=27) with high-grade glioma or ependymoma. The maximum tolerated dose (MTD) normalized for body surface area (BSA) was lower in pediatric patients compared to adults. Sunitinib was poorly tolerated in pediatric patients. The occurrence of dose-limiting cardiotoxicity prompted an amendment of the NCT00387920 study to exclude patients with previous exposure to anthracyclines or cardiac radiation. No responses were reported in patients in either of the trials.
Apparent clearance and volume of distribution normalized for BSA for sunitinib and its active major metabolite were lower in pediatrics as compared to adults.
The effect on open tibial growth plates in pediatric patients who received SUTENT has not been adequately studied. See Juvenile Animal Toxicity Data below.
8.5 Geriatric Use
Of the 7527 patients with GIST, RCC (advanced and adjuvant), or pNET who received SUTENT, 32% were 65 years and older, and 7% were 75 years and older. Patients aged 65 years of age and older had a higher incidence of Grade 3 or 4 adverse reactions (67%) than younger patients (60%).
In the GIST study, 73 (30%) of the patients who received SUTENT were 65 years and older. In the mRCC study, 152 (41%) of patients who received SUTENT were 65 years and older. No overall differences in safety or effectiveness were observed between these patients and younger patients.
In the pNET study, 22 (27%) of the patients who received SUTENT were 65 years and older. Clinical studies of SUTENT did not include sufficient numbers of patients with pNET to determine if patients 65 years of age and older respond differently than younger patients.
5.1 Hepatotoxicity
SUTENT can cause severe hepatotoxicity, resulting in liver failure or death. In the pooled safety population, liver failure occurred in <1% of patients in clinical trials. Liver failure include jaundiced, elevated transaminases and/or hyperbilirubinemia in conjunction with encephalopathy, coagulopathy, and/or renal failure.
Monitor liver function tests (alanine aminotransferase [ALT], aspartate aminotransferase [AST], and bilirubin) at baseline, during each cycle, and as clinically indicated. Interrupt SUTENT for Grade 3 hepatotoxicity until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose.
Discontinue SUTENT in patients with Grade 4 hepatotoxicity, in patients without resolution of Grade 3 hepatotoxicity, in patients who subsequently experience severe changes in liver function tests and in patients who have other signs and symptoms of liver failure. Safety in patients with ALT or AST >2.5 × upper limit of normal (ULN) or with >5 × ULN and liver metastases has not been established.
4 Contraindications
None.
6 Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling.
-
•Hepatotoxicity [see Warnings and Precautions (5.1)]
-
•Cardiovascular Events [see Warnings and Precautions (5.2)]
-
•QT Interval Prolongation and Torsade de Pointes [see Warnings and Precautions (5.3)]
-
•Hypertension [see Warnings and Precautions (5.4)]
-
•Hemorrhagic Events [see Warnings and Precautions (5.5)]
-
•Tumor Lysis Syndrome [see Warnings and Precautions (5.6)]
-
•Thrombotic Microangiopathy [see Warnings and Precautions (5.7)]
-
•Proteinuria [see Warnings and Precautions (5.8)]
-
•Dermatologic Toxicities [see Warnings and Precautions (5.9)]
-
•Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.10)]
-
•Thyroid Dysfunction [see Warnings and Precautions (5.11)]
-
•Hypoglycemia [see Warnings and Precautions (5.12)]
-
•Osteonecrosis of the Jaw [see Warnings and Precautions (5.13)]
-
•Impaired Wound Healing [see Warnings and Precautions (5.14)]
7 Drug Interactions
8.7 Renal Impairment
No dose adjustment is recommended in patients with mild (CLcr 50 to 80 mL/min), moderate (CLcr 30 to <50 mL/min), or severe (CLcr <30 mL/min) renal impairment who are not on dialysis [see Clinical Pharmacology (12.3)].
No dose adjustment is recommended for patients with end-stage renal disease (ESRD) on hemodialysis [see Clinical Pharmacology (12.3)].
12.3 Pharmacokinetics
The pharmacokinetics of sunitinib and sunitinib malate have been evaluated in healthy subjects and in patients with solid tumors.
Sunitinib AUC and Cmax increase proportionately over a dose range of 25 mg to 100 mg (0.5 to 2 times the approved RDD of 50 mg). The pharmacokinetics were similar in healthy subjects and in patients with a solid tumor, including patients with GIST and RCC. No significant changes in the pharmacokinetics of sunitinib or the primary active metabolite were observed with repeated daily administration or with repeated cycles. With repeated daily administration, sunitinib accumulates 3- to 4-fold while the primary metabolite accumulates 7- to 10-fold. Steady-state concentrations of sunitinib and its primary active metabolite are achieved within 10 to 14 days. By Day 14, combined plasma concentrations of sunitinib and its active metabolite ranged from 63 to 101 ng/mL.
8.6 Hepatic Impairment
No dose adjustment is required in patients with mild or moderate (Child-Pugh Class A or B) hepatic impairment [see Clinical Pharmacology (12.3)]. SUTENT was not studied in patients with severe (Child-Pugh Class C) hepatic impairment.
1 Indications and Usage
SUTENT is a kinase inhibitor indicated for:
-
•treatment of adult patients with gastrointestinal stromal tumor (GIST) after disease progression on or intolerance to imatinib mesylate. (1.1)
-
•treatment of adult patients with advanced renal cell carcinoma (RCC). (1.2)
-
•adjuvant treatment of adult patients at high risk of recurrent RCC following nephrectomy. (1.3)
-
•treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNET) in adult patients with unresectable locally advanced or metastatic disease. (1.4)
Warning: Hepatotoxicity
Hepatotoxicity may be severe, and in some cases, fatal. Monitor hepatic function and interrupt, dose reduce, or discontinue SUTENT as recommended [see Warnings and Precautions (5.1)].
12.1 Mechanism of Action
Sunitinib is a small molecule that inhibits multiple receptor tyrosine kinases (RTKs), some of which are implicated in tumor growth, pathologic angiogenesis, and metastatic progression of cancer. Sunitinib was evaluated for its inhibitory activity against a variety of kinases (>80 kinases) and was identified as an inhibitor of platelet-derived growth factor receptors (PDGFRα and PDGFRβ), vascular endothelial growth factor receptors (VEGFR1, VEGFR2, and VEGFR3), stem cell factor receptor (KIT), Fms-like tyrosine kinase-3 (FLT3), colony stimulating factor receptor Type 1 (CSF-1R), and the glial cell-line derived neurotrophic factor receptor (RET). Sunitinib inhibition of the activity of these RTKs has been demonstrated in biochemical and cellular assays, and inhibition of function has been demonstrated in cell proliferation assays. The primary metabolite exhibits similar potency compared to sunitinib in biochemical and cellular assays.
Sunitinib inhibited the phosphorylation of multiple RTKs (PDGFRβ, VEGFR2, KIT) in tumor xenografts expressing RTK targets in vivo and demonstrated inhibition of tumor growth or tumor regression and/or inhibited metastases in some experimental models of cancer. Sunitinib demonstrated the ability to inhibit growth of tumor cells expressing dysregulated target RTKs (PDGFR, RET, or KIT) in vitro and to inhibit PDGFRβ- and VEGFR2-dependent tumor angiogenesis in vivo.
5.11 Thyroid Dysfunction
Hyperthyroidism, some followed by hypothyroidism, have been reported in clinical trials and through postmarketing experience of SUTENT.
Monitor thyroid function at baseline, periodically during treatment and as clinically indicated. Monitor patients closely for signs and symptoms of thyroid dysfunction, including hypothyroidism, hyperthyroidism, and thyroiditis, during treatment with SUTENT. Initiate and/or adjust therapies for thyroid dysfunction as appropriate.
13 Nonclinical Toxicology
5.2 Cardiovascular Events
Cardiovascular events, including heart failure, cardiomyopathy, myocardial ischemia, and myocardial infarction, some of which were fatal, have been reported.
In pooled safety population, 3% of patients experienced heart failure; 71% of the patients with heart failure were reported as recovered. Fatal cardiac failure was reported in <1% of patients.
In the adjuvant treatment of RCC study, 11 patients experienced Grade 2 decreased ejection fraction (left ventricular ejection fraction [LVEF] 40% to 50% and a 10% to 19% decrease from baseline). In 3 of these 11 patients, the ejection fractions arm did not return to ≥50% or baseline by the time of last measurement. No patients who received SUTENT were diagnosed with CHF.
Patients who presented with cardiac events within 12 months prior to SUTENT administration, such as myocardial infarction (including severe/unstable angina), coronary/peripheral artery bypass graft, symptomatic CHF, cerebrovascular accident or transient ischemic attack, or pulmonary embolism were excluded from SUTENT clinical studies. Patients with prior anthracycline use or cardiac radiation were also excluded from some studies. It is unknown whether patients with these concomitant conditions may be at a higher risk of developing left ventricular dysfunction.
Consider monitoring LVEF at baseline and periodically as clinically indicated. Carefully monitor patients for clinical signs and symptoms of congestive heart failure (CHF). Discontinue SUTENT in patients who experience clinical manifestations of CHF. Interrupt SUTENT and/or reduce the dose in patients without clinical evidence of CHF who have an ejection fraction of greater than 20% but less than 50% below baseline or below the lower limit of normal if baseline ejection fraction was not obtained.
5 Warnings and Precautions
-
•Hepatotoxicity: Fatal liver failure has been observed. Monitor liver function tests at baseline, during each cycle, and as clinically indicated. Interrupt SUTENT for Grade 3 hepatotoxicity until resolution to Grade ≤1 or baseline and resume SUTENT at a reduced dose; discontinue if no resolution. Discontinue SUTENT in patients with Grade 4 hepatoxicity, in patients who have subsequent severe changes in liver function tests or other signs and symptoms of liver failure. (2.4, 5.1)
-
•Cardiovascular Events: Myocardial ischemia, myocardial infarction, heart failure, cardiomyopathy, and decreased left ventricular ejection fraction (LVEF) to below the lower limit of normal including death have occurred. Monitor for signs and symptoms of congestive heart failure and consider monitoring LVEF at baseline and periodically during treatment. Discontinue SUTENT for clinical manifestations of congestive heart failure. Interrupt and/or dose reduce for decreased LVEF. (5.2)
-
•QT Interval Prolongation and Torsade de Pointes: Monitor patients at higher risk for developing QT interval prolongation. Consider monitoring of electrocardiograms and electrolytes. (5.3)
-
•Hypertension: Monitor blood pressure at baseline and as clinically indicated. Initiate and/or adjust antihypertensive therapy as appropriate. Interrupt SUTENT for Grade 3 hypertension until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose. Discontinue SUTENT in patients who develop Grade 4 hypertension. (5.4)
-
•Hemorrhagic Events: Tumor-related hemorrhage and viscus perforation (both with fatal events) have occurred. Perform serial complete blood counts and physical examinations. Interrupt SUTENT for Grade 3 or 4 hemorrhagic events until resolution to Grade ≤1 or baseline, then resume at a reduced dose; discontinue if no resolution. (5.5)
-
•Tumor Lysis Syndrome (TLS): TLS (some fatal) has been reported primarily in patients with RCC and GIST. Monitor these patients and treat as clinically indicated. (5.6)
-
•Thrombotic microangiopathy (TMA): TMA, including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome, sometimes leading to renal failure or a fatal outcome, has been reported. Discontinue SUTENT for TMA. (5.7)
-
•Proteinuria: Renal failure or a fatal outcome has occurred. Monitor urine protein. Interrupt treatment for 24-hour urine protein of 3 or more grams. Discontinue for repeat episodes of 24-hour urine protein of 3 or more grams despite dose reductions or nephrotic syndrome. (5.8)
-
•Dermatologic Toxicities: Necrotizing fasciitis, erythema multiforme, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) (some fatal) have occurred. Discontinue SUTENT for these events. (5.9)
-
•Reversible Posterior Leukoencephalopathy Syndrome (RPLS): RPLS (some fatal) has been reported. Monitor for signs and symptoms of RPLS. Withhold SUTENT until resolution. (5.10)
-
•Thyroid Dysfunction: Monitor thyroid function at baseline, periodically during treatment, and as clinically indicated. Initiate and/or adjust therapy for thyroid dysfunction as appropriate. (5.11)
-
•Hypoglycemia: Check blood glucose levels regularly and assess if antidiabetic drug dose modifications are required. (5.12)
-
•Osteonecrosis of the Jaw (ONJ): Withhold SUTENT for at least 3 weeks prior to invasive dental procedure and for development of ONJ until complete resolution. (5.13)
-
•Impaired Wound Healing: Withhold SUTENT for at least 3 weeks prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of SUTENT after resolution of wound healing complications has not been established. (5.14)
-
•Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.15, 8.1, 8.3)
5.15 Embryo Fetal Toxicity
Based on findings from animal studies and its mechanism of action, SUTENT can cause fetal harm when administered to pregnant woman. Administration of sunitinib to pregnant rats and rabbits during the period of organogenesis resulted in teratogenicity at approximately 5.5 and 0.3 times the combined systemic exposure [combined area under the curve (AUC) of sunitinib plus its active metabolite] in patients administered the recommended daily dose (RDD) of 50 mg, respectively.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with SUTENT and for 4 weeks following the final dose [see Use in Specific Populations (8.1, 8.3)].
2 Dosage and Administration
GIST and Advanced RCC:
-
•The recommended dosage is 50 mg orally once daily for the first 4 weeks of each 6-week cycle (Schedule 4/2). (2.1)
Adjuvant Treatment of RCC:
-
•The recommended dosage is 50 mg orally once daily for the first 4 weeks of a 6-week cycle (Schedule 4/2) for a maximum of 9 cycles. (2.2)
pNET:
-
•The recommended dosage is 37.5 mg orally once daily. (2.3)
5.14 Impaired Wound Healing
Impaired wound healing has been reported in patients who received SUTENT [see Adverse Reactions (6.2)].
Withhold SUTENT for at least 3 weeks prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of SUTENT after resolution of wound healing complications has not been established.
5.9 Dermatologic Toxicities
Severe cutaneous adverse reactions have been reported, including erythema multiforme (EM), Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN), some of which were fatal. Permanently discontinue SUTENT for these severe cutaneous adverse reactions.
Necrotizing fasciitis, including fatal cases, has been reported in patients treated with SUTENT, including of the perineum and secondary to fistula formation. Discontinue SUTENT in patients who develop necrotizing fasciitis.
3 Dosage Forms and Strengths
Capsules, hard gelatin:
-
•12.5 mg sunitinib: orange cap and orange body, printed with white ink "Pfizer" on the cap and "STN 12.5 mg" on the body.
-
•25 mg sunitinib: caramel cap and orange body, printed with white ink "Pfizer" on the cap and "STN 25 mg" on the body.
-
•37.5 mg sunitinib: yellow cap and yellow body, printed with black ink "Pfizer" on the cap and "STN 37.5 mg" on the body.
-
•50 mg sunitinib: caramel top and caramel body, printed with white ink "Pfizer" on the cap and "STN 50 mg" on the body.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of SUTENT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
•Blood and lymphatic system disorders: hemorrhage associated with thrombocytopeniaincluding some fatalities.
-
•Gastrointestinal disorders: esophagitis.
-
•Hepatobiliary disorders: cholecystitis, particularly acalculous cholecystitis.
-
•Immune system disorders: hypersensitivity reactions, including angioedema.
-
•Infections and infestations: serious infection (with or without neutropenia). The infections most commonly observed with SUTENT include respiratory, urinary tract, skin infections, and sepsis/septic shock.
-
•Musculoskeletal and connective tissue disorders: fistula formation, sometimes associated with tumor necrosis and/or regression; myopathy and/or rhabdomyolysis with or without acute renal failure.
-
•Renal and urinary disorders: renal impairment and/or failure.
-
•Respiratory disorders: pulmonary embolism, pleural effusion.
-
•Skin and subcutaneous tissue disorders: pyoderma gangrenosum, including positive de-challenges.
-
•Vascular disorders: arterial (including aortic) aneurysms, dissections, and rupture; arterial thromboembolic events. The most frequent events included cerebrovascular accident, transient ischemic attack, and cerebral infarction.
-
•General disorders and administration site conditions: impaired wound healing.
8 Use in Specific Populations
-
•Lactation: Advise not to breastfeed. (8.2)
5.6 Tumor Lysis Syndrome (tls)
Tumor Lysis Syndrome (TLS), some fatal, occurred in clinical trials and has been reported in postmarketing experience, primarily in patients with RCC or GIST. Patients generally at risk of TLS are those with high tumor burden prior to treatment. Monitor these patients for TLS and manage as appropriate.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the Warnings and Precautions reflect exposure to SUTENT in 7527 patients with GIST, RCC (advanced and adjuvant), or pNET. In this pooled safety population, the most common adverse reactions (≥25%) were fatigue/asthenia, diarrhea, mucositis/stomatitis, nausea, decreased appetite/anorexia, vomiting, abdominal pain, hand-foot syndrome, hypertension, bleeding events, dysgeusia/altered taste, dyspepsia, and thrombocytopenia.
2.3 Recommended Dosage for Pnet
The recommended dosage of SUTENT for pancreatic neuroendocrine tumors (pNET) is 37.5 mg taken orally once daily until disease progression or unacceptable toxicity. SUTENT may be taken with or without food.
1.2 Advanced Renal Cell Carcinoma
SUTENT is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC).
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
1.1 Gastrointestinal Stromal Tumor
SUTENT is indicated for the treatment of adult patients with gastrointestinal stromal tumor (GIST) after disease progression on or intolerance to imatinib mesylate.
7.2 Drugs That Prolong Qt Interval
SUTENT is associated with QTc interval prolongation [see Warnings and Precautions (5.3), Clinical Pharmacology (12.2)]. Monitor the QT interval with ECGs more frequently in patients who require treatment with concomitant medications known to prolong the QT interval.
5.13 Osteonecrosis of the Jaw (onj)
Osteonecrosis of the Jaw (ONJ) occurred in patients treated with SUTENT. Concomitant exposure to other risk factors, such as bisphosphonates or dental disease/invasive dental procedures, may increase the risk of ONJ. Perform an oral examination prior to initiation of SUTENT and periodically during SUTENT therapy. Advise patients regarding good oral hygiene practices. Withhold SUTENT treatment for at least 3 weeks prior to scheduled dental surgery or invasive dental procedures, if possible. Withhold SUTENT for development of ONJ until complete resolution. The safety of resumption of SUTENT after resolution of osteonecrosis of the jaw has not been established.
16 How Supplied/storage and Handling
SUTENT 12.5 mg capsules are supplied as hard gelatin capsule with orange cap and orange body, printed with white ink "Pfizer" on the cap, "STN 12.5 mg" on the body:
Bottles of 28 capsules: NDC 0069-0550-38
SUTENT 25 mg capsules are supplied as hard gelatin capsule with caramel cap and orange body, printed with white ink "Pfizer" on the cap, "STN 25 mg" on the body:
Bottles of 28 capsules: NDC 0069-0770-38
SUTENT 37.5 mg capsules are supplied as hard gelatin capsule with yellow cap and yellow body, printed with black ink "Pfizer" on the cap, "STN 37.5 mg" on the body:
Bottles of 28 capsules: NDC 0069-0830-38
SUTENT 50 mg capsules are supplied as hard gelatin capsule with caramel cap and caramel body, printed with white ink "Pfizer" on the cap, "STN 50 mg" on the body:
Bottles of 28 capsules: NDC 0069-0980-38
5.7 Thrombotic Microangiopathy (tma)
Thrombotic Microangiopathy (TMA), including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome, sometimes leading to renal failure or a fatal outcome, occurred in clinical trials and in postmarketing experience of SUTENT as monotherapy and administered in combination with bevacizumab. SUTENT is not approved for use in combination with bevacizumab.
Discontinue SUTENT in patients developing TMA. Reversal of the effects of TMA has been observed after SUTENT was discontinued.
14.3 Pancreatic Neuroendocrine Tumors
Study 6 (NCT#00428597) was a multi-center, international, randomized, double-blind, placebo-controlled study of single-agent SUTENT conducted in patients with unresectable pNET. Patients were required to have documented RECIST-defined disease progression within the prior 12 months and were randomized (1:1) to receive either 37.5 mg SUTENT (N=86) or placebo (N=85) once daily without a scheduled off-treatment period. The primary objective was to compare PFS in patients receiving SUTENT versus patients receiving placebo. Other endpoints included OS, ORR, and safety. Use of somatostatin analogs was allowed in the study.
Demographics were comparable between the SUTENT and placebo groups. Additionally, 49% of SUTENT patients had nonfunctioning tumors vs 52% of placebo patients, and 92% patients in both arms had liver metastases. A total of 66% of SUTENT patients received prior systemic therapy compared with 72% of placebo patients and 35% of SUTENT patients had received somatostatin analogs compared with 38% of placebo patients. Patients were treated until disease progression or withdrawal from the study. Upon disease progression or study closure, patients were offered access to SUTENT in a separate extension study.
As recommended by the Independent Data Monitoring Committee, the study was terminated prematurely prior to the prespecified interim analysis. This may have led to an overestimate of the magnitude of PFS effect. A clinically significant improvement for SUTENT over placebo in PFS was seen by both investigator and independent assessment. A hazard ratio favoring SUTENT was observed in all subgroups of baseline characteristics evaluated. OS data were not mature at the time of the analysis. There were 9 deaths in the SUTENT arm and 21 deaths in the placebo arm. A statistically significant difference in ORR favoring SUTENT over placebo was observed. Efficacy results are summarized in Table 15 and the Kaplan-Meier curve for PFS is in Figure 4.
| Abbreviations: CI=confidence interval; HR=hazard ratio; N=number of patients; NA=not applicable; pNET=pancreatic neuroendocrine tumors. | ||||
|
Efficacy Parameter |
SUTENT
|
Placebo
|
p-value |
HR
|
|
Progression-free survival |
10.2 |
5.4 |
0.000146 2-sided unstratified log-rank test.
|
0.427 |
|
Objective response rate |
9.3 |
0 |
0.0066 Fisher's Exact test.
|
NA |
Figure 4. Kaplan-Meier Curve of PFS in the pNET Study 6
Abbreviations: CI=confidence interval; N=number of patients; PFS=progression-free survival; pNET=pancreatic neuroendocrine tumors.
1.4 Advanced Pancreatic Neuroendocrine Tumors
SUTENT is indicated for the treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNET) in adult patients with unresectable locally advanced or metastatic disease.
5.5 Hemorrhagic Events and Viscus Perforation
Hemorrhagic events, some of which were fatal, have involved the gastrointestinal tract, respiratory tract, tumor, urinary tract, and brain. In the pooled safety population, 30% of patients experienced hemorrhagic events, including Grade 3 or 4 in 4.2% of patients. Epistaxis was the most common hemorrhagic event and gastrointestinal hemorrhage was the most common Grade 3–5 event.
Tumor-related hemorrhage was observed in patients treated with SUTENT. These events may occur suddenly, and in the case of pulmonary tumors, may present as severe and life-threatening hemoptysis or pulmonary hemorrhage. Pulmonary hemorrhage, some with a fatal outcome, was observed in patients treated with SUTENT for metastatic RCC, GIST, and metastatic lung cancer. SUTENT is not approved for use in patients with lung cancer.
Serious, sometimes fatal, gastrointestinal complications including gastrointestinal perforation, have been reported in patients with intra-abdominal malignancies treated with SUTENT.
Include serial complete blood counts (CBCs) and physical examinations with the clinical assessment of hemorrhagic events. Interrupt SUTENT for Grade 3 or 4 hemorrhagic events until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose.
Discontinue SUTENT in patients without resolution of Grade 3 or 4 hemorrhagic events.
1.3 Adjuvant Treatment of Renal Cell Carcinoma
SUTENT is indicated for the adjuvant treatment of adult patients at high risk of recurrent RCC following nephrectomy.
2.4 Dosage Modifications for Adverse Reactions
To manage adverse reactions, the recommended dosage modifications are provided in Table 1. Table 2 provides the recommended dosage reductions of SUTENT for adverse reactions.
|
Indications |
GIST |
RCC |
pNET |
|
|
Advanced RCC |
Adjuvant RCC |
|||
|
First dose reduction |
37.5 mg once daily |
37.5 mg once daily |
37.5 mg once daily |
25 mg once daily |
|
Second dose reduction |
25 mg once daily |
25 mg once daily |
NA |
NA |
|
Adverse Reaction |
Severity |
Dosage Modifications for SUTENT |
|
Hepatotoxicity [see Warnings and Precautions (5.1)] |
Grade 3 |
|
|
Grade 4 |
|
|
|
Cardiovascular events [see Warnings and Precautions (5.2)] |
Asymptomatic cardiomyopathy (left ventricular ejection fraction greater than 20% but less than 50% below baseline or below the lower limit of normal if baseline was not obtained) |
|
|
Clinically manifested congestive heart failure (CHF) |
|
|
|
Hypertension [see Warnings and Precautions (5.4)] |
Grade 3 |
|
|
Grade 4 |
|
|
|
Hemorrhagic events [see Warnings and Precautions (5.5)] |
Grade 3 or 4 |
|
|
Thrombotic microangiopathy [see Warnings and Precautions (5.7)] |
Any Grade |
|
|
Proteinuria or Nephrotic syndrome [see Warnings and Precautions (5.8)] |
3 or more grams proteinuria in 24 hours in the absence of nephrotic syndrome |
|
|
Nephrotic syndrome or recurrent proteinuria of 3 or more grams per 24 hours despite dose reductions |
|
|
|
Dermatological toxicities Erythema multiforme (EM), Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), Necrotizing fasciitis [see Warnings and Precautions (5.9)] |
Any Grade |
|
|
Reversible posterior leukoencephalopathy syndrome [see Warnings and Precautions (5.10)] |
Any Grade |
|
|
Osteonecrosis of the jaw [see Warnings and Precautions (5.13)] |
Any Grade |
|
|
Impaired wound healing [see Warnings and Precautions (5.14)] |
Any Grade |
|
8.3 Females and Males of Reproductive Potential
SUTENT can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
2.1 Recommended Dosage for Gist and Advanced Rcc
The recommended dosage of SUTENT for gastrointestinal stromal tumor (GIST) and advanced renal cell carcinoma (RCC) is 50 mg taken orally once daily, on a schedule of 4 weeks on treatment followed by 2 weeks off (Schedule 4/2) until disease progression or unacceptable toxicity. SUTENT may be taken with or without food.
5.3 Qt Interval Prolongation and Torsade De Pointes
SUTENT can cause QT interval prolongation in a dose-dependent manner, which may lead to an increased risk for ventricular arrhythmias including Torsade de Pointes. Torsade de Pointes was observed in <0.1% of patients.
Monitor patients who are at higher risk of developing QT interval prolongation, including patients with a history of QT interval prolongation, patients who are taking antiarrhythmics, or patients with relevant pre-existing cardiac disease, bradycardia, or electrolyte disturbances. Consider periodic monitoring of electrocardiograms and electrolytes (i.e., magnesium, potassium) during treatment with SUTENT.
Monitor QT interval more frequently when SUTENT is concomitantly administered with strong CYP3A4 inhibitors or drugs known to prolong QT interval. Consider dose reducing SUTENT [see Dosage and Administration (2.5), Drug Interactions (7.2)].
2.2 Recommended Dosage for Adjuvant Treatment of Rcc
The recommended dosage of SUTENT for the adjuvant treatment of RCC is 50 mg taken orally once daily, on a schedule of 4 weeks on treatment followed by 2 weeks off (Schedule 4/2), for nine 6-week cycles. SUTENT may be taken with or without food.
Principal Display Panel 50 Mg Capsule Bottle Label
PROFESSIONAL SAMPLE – NOT FOR SALE
ALWAYS DISPENSE WITH MEDICATION GUIDE
Pfizer
NDC 63539-019-01
Sutent®
(sunitinib malate)
50 mg*
Capsules
14 Capsules
Rx only
Principal Display Panel 12.5 Mg Capsule Bottle Label
PROFESSIONAL SAMPLE – NOT FOR SALE
ALWAYS DISPENSE WITH MEDICATION GUIDE
Pfizer
NDC 63539-017-01
Sutent®
(sunitinib malate)
12.5 mg*
Capsules
28 Capsules
Rx only
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of sunitinib has been evaluated in 2 species: rasH2 transgenic mice and Sprague-Dawley rats. There were similar positive findings in both species. In rasH2 transgenic mice, gastroduodenal carcinomas and/or gastric mucosal hyperplasia, as well as an increased incidence of background hemangiosarcomas were observed at sunitinib daily doses of ≥25 mg/kg/day in studies of 1 or 6 months duration. No proliferative changes were observed in rasH2 transgenic mice at 8 mg/kg/day. Similarly, in a 2-year rat carcinogenicity study, administration of sunitinib in 28-day cycles followed by 7-day dose-free periods resulted in findings of duodenal carcinoma at doses as low as 1 mg/kg/day [approximately 0.9 times the combined AUC (combined systemic exposure of sunitinib plus its active metabolite) in patients administered the RDD of 50 mg]. At the high dose of 3 mg/kg/day (approximately 8 times the combined AUC in patients administered the RDD of 50 mg), the incidence of duodenal tumors was increased and was accompanied by findings of gastric mucous cell hyperplasia and by an increased incidence of pheochromocytoma and hyperplasia of the adrenal gland.
Sunitinib did not cause genetic damage when tested in in vitro assays [bacterial mutation (Ames test), human lymphocyte chromosome aberration] and an in vivo rat bone marrow micronucleus test.
In a female fertility and early embryonic development study, female rats were administered oral sunitinib (0.5, 1.5, 5 mg/kg/day) for 21 days prior to mating and for 7 days after mating. Preimplantation loss was observed in females administered 5 mg/kg/day (approximately 5 times the combined AUC in patients administered the RDD of 50 mg). No adverse effects on fertility were observed at doses ≤1.5 mg/kg/day (approximately equal to the combined AUC in patients administered the RDD of 50 mg). In addition, effects on the female reproductive system were identified in a 3-month oral repeat-dose monkey study (2, 6, 12 mg/kg/day). Ovarian changes (decreased follicular development) were noted at 12 mg/kg/day (approximately 5 times the combined AUC in patients administered the RDD of 50 mg), while uterine changes (endometrial atrophy) were noted at ≥2 mg/kg/day (approximately 0.4 times the combined AUC in patients administered the RDD of 50 mg). With the addition of vaginal atrophy, the uterine and ovarian effects were reproduced at 6 mg/kg/day (approximately 0.8 times the combined AUC in patients administered the RDD of 50 mg) in a 9-month monkey study (0.3, 1.5, and 6 mg/kg/day administered daily for 28 days followed by a 14-day respite).
In a male fertility study, no reproductive effects were observed in male rats dosed with 1, 3, or 10 mg/kg/day oral sunitinib for 58 days prior to mating with untreated females. Fertility, copulation, conception indices, and sperm evaluation (morphology, concentration, and motility) were unaffected by sunitinib at doses ≤10 mg/kg/day (approximately ≥26 times the combined AUC in patients administered the RDD of 50 mg).
5.10 Reversible Posterior Leukoencephalopathy Syndrome (rpls)
Reversible posterior leukoencephalopathy syndrome (RPLS) has been reported in <1% of patients, some of which were fatal. Patients can present with hypertension, headache, decreased alertness, altered mental functioning, and visual loss, including cortical blindness. Magnetic resonance imaging is necessary to confirm the diagnosis. Discontinue SUTENT in patients developing RPLS.
2.6 Dosage Modification for End Stage Renal Disease Patients On Hemodialysis
No starting dose adjustment is required in patients with end-stage renal disease (ESRD) on hemodialysis. However, given the decreased exposure compared to patients with normal renal function, subsequent doses may be increased gradually up to 2-fold based on safety and tolerability [see Clinical Pharmacology (12.3)].
Structured Label Content
Section 42229-5 (42229-5)
Strong CYP3A4 Inhibitors
Select an alternate concomitant medication with no or minimal enzyme inhibition potential. If coadministration of SUTENT with a strong CYP3A4 inhibitor cannot be avoided, consider a dose reduction for SUTENT to a minimum dosage as follows [see Drug Interactions (7.1)]:
-
•GIST and RCC: 37.5 mg orally once daily, on a schedule of 4 weeks on treatment followed by 2 weeks off (Schedule 4/2)
-
•pNET: 25 mg orally once daily
Section 42231-1 (42231-1)
| This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: October 2025 | ||
|
MEDICATION GUIDE
|
||
|
What is the most important information I should know about SUTENT?
Your healthcare provider should do blood tests to check your liver function before you start taking and during treatment with SUTENT. Your healthcare provider may temporarily stop, reduce your dose, or permanently stop treatment with SUTENT if you develop liver problems. |
||
|
What is SUTENT?
It is not known if SUTENT is safe and effective in children. |
||
|
Before taking SUTENT tell your healthcare provider about all of your medical conditions, including if you:
Tell all of your healthcare providers and dentists that you are taking SUTENT. They should talk to the healthcare provider who prescribed SUTENT for you, before you have any surgery, or medical or dental procedure. |
||
|
How should I take SUTENT?
|
||
|
What are possible side effects of SUTENT?
|
||
|
|
|
|
||
|
|
|
|
Your healthcare provider may prescribe medicine for you to treat high blood pressure, if needed. |
||
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
Your healthcare provider may temporarily stop, reduce your dose, or permanently stop treatment with SUTENT if you develop serious side effects.
|
||
|
|
|
|
The medicine in SUTENT is yellow, and it may make your skin look yellow. Your skin and hair may get lighter in color. SUTENT may also cause other skin problems including: dryness, thickness or cracking of the skin. |
||
|
How do I store SUTENT?
Keep SUTENT and all medicines out of the reach of children. |
||
|
General information about the safe and effective use of SUTENT.
|
||
|
What are the ingredients in SUTENT?
LAB-0361-13.0 |
Section 44425-7 (44425-7)
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
10 Overdosage (10 OVERDOSAGE)
Treatment of overdose with SUTENT should consist of general supportive measures. There is no specific antidote for overdosage with SUTENT. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage. Cases of accidental overdose have been reported; these cases were associated with adverse reactions consistent with the known safety profile of SUTENT, or without adverse reactions. In nonclinical studies, mortality was observed following as few as 5 daily doses of 500 mg/kg (3000 mg/m2) in rats. At this dose, signs of toxicity included impaired muscle coordination, head shakes, hypoactivity, ocular discharge, piloerection, and gastrointestinal distress. Mortality and similar signs of toxicity were observed at lower doses when administered for longer durations.
8.2 Lactation
There is no information regarding the presence of sunitinib and its metabolites in human milk. Sunitinib and its metabolites were excreted in rat milk at concentrations up to 12-fold higher than in plasma (see Data). Because of the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with SUTENT and for at least 4 weeks after the last dose.
11 Description (11 DESCRIPTION)
Sunitinib is a kinase inhibitor present in SUTENT capsules as the malate salt. Sunitinib malate is described chemically as (2S)-2-hydroxybutanedoic acid with N-[2-(diethylamino)ethyl]-5-[(Z)-(5-fluoro-1,2-dihydro-2-oxo-3H-indol-3-ylidine)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide (1:1). The molecular formula is C22H27FN4O2 ∙ C4H6O5 and the molecular weight is 532.6 Daltons. The chemical structure of sunitinib malate is:
Sunitinib malate is a yellow to orange powder with a pKa of 8.95. The solubility of sunitinib malate in aqueous media over the range pH 1.2 to pH 6.8 is in excess of 25 mg/mL. The log of the distribution coefficient (octanol/water) at pH 7 is 5.2.
SUTENT (sunitinib malate) capsules are supplied as printed hard shell capsules containing 12.5 mg, 25 mg, 37.5 mg or 50 mg of sunitinib (equivalent to 16.7 mg, 33.4 mg, 50.1 mg, or 66.8 mg of sunitinib malate, respectively). The capsules contain the following inactive ingredients: croscarmellose sodium, magnesium stearate, mannitol, and povidone (K-25). The orange gelatin capsule shells contain titanium dioxide and red iron oxide; the caramel gelatin capsule shells contain titanium dioxide, red iron oxide, yellow iron oxide, and black iron oxide; and the yellow gelatin capsule shells contain titanium dioxide and yellow iron oxide. The white printing ink contains shellac, propylene glycol, sodium hydroxide, povidone, and titanium dioxide and the black printing ink contains shellac, propylene glycol, potassium hydroxide, and black iron oxide.
5.8 Proteinuria
Proteinuria and nephrotic syndrome have been reported. Some of these cases have resulted in renal failure and fatal outcomes.
Monitor patients for the development or worsening of proteinuria. Perform baseline and periodic urinalyses during treatment, with follow up measurement of 24-hour urine protein as clinically indicated. Interrupt SUTENT and dose reduce for 24-hour urine protein of 3 or more grams. Discontinue SUTENT for patients with nephrotic syndrome or repeat episodes of 24-hour urine protein of 3 or more grams despite dose reductions. The safety of continued SUTENT treatment in patients with moderate to severe proteinuria has not been evaluated.
5.4 Hypertension
In the pooled safety population, 29% of patients experienced hypertension. Grade 3 hypertension was reported in 7% of patients, and Grade 4 hypertension was reported in 0.2%.
Monitor blood pressure at baseline and as clinically indicated. Initiate and/or adjust antihypertensive therapy as appropriate. In cases of Grade 3 hypertension, withhold SUTENT until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose. Discontinue SUTENT in patients with who develop Grade 4 hypertension.
5.12 Hypoglycemia
SUTENT can result in symptomatic hypoglycemia, which may lead to loss of consciousness, or require hospitalization. In the pooled safety population, hypoglycemia occurred in 2% of the patients treated with SUTENT. Hypoglycemia has occurred in clinical trials in 2% of the patients treated with SUTENT for advanced RCC (Study 3) and GIST (Study 1) (n=577) and in approximately 10% of the patients treated with SUTENT for pNET (Study 6) (n=83). For patients being treated with SUTENT for pNET, pre-existing abnormalities in glucose homeostasis were not present in all patients who experienced hypoglycemia. Reductions in blood glucose levels may be worse in patients with diabetes.
Check blood glucose levels at baseline, regularly during treatment, as clinically indicated and after discontinuation of SUTENT. In patients with diabetes, assess if antidiabetic therapies need to be adjusted to minimize the risk of hypoglycemia.
8.4 Pediatric Use
The safety and effectiveness of SUTENT in pediatric patients have not been established. Safety and pharmacokinetics of sunitinib were assessed in an open-label study (NCT00387920) in pediatric patients 2 years to <17 years of age (n=29) with refractory solid tumors. In addition, efficacy, safety and pharmacokinetics of sunitinib was assessed in another open-label study (NCT01462695) in pediatric patients 2 years to <17 years of age (n=27) with high-grade glioma or ependymoma. The maximum tolerated dose (MTD) normalized for body surface area (BSA) was lower in pediatric patients compared to adults. Sunitinib was poorly tolerated in pediatric patients. The occurrence of dose-limiting cardiotoxicity prompted an amendment of the NCT00387920 study to exclude patients with previous exposure to anthracyclines or cardiac radiation. No responses were reported in patients in either of the trials.
Apparent clearance and volume of distribution normalized for BSA for sunitinib and its active major metabolite were lower in pediatrics as compared to adults.
The effect on open tibial growth plates in pediatric patients who received SUTENT has not been adequately studied. See Juvenile Animal Toxicity Data below.
8.5 Geriatric Use
Of the 7527 patients with GIST, RCC (advanced and adjuvant), or pNET who received SUTENT, 32% were 65 years and older, and 7% were 75 years and older. Patients aged 65 years of age and older had a higher incidence of Grade 3 or 4 adverse reactions (67%) than younger patients (60%).
In the GIST study, 73 (30%) of the patients who received SUTENT were 65 years and older. In the mRCC study, 152 (41%) of patients who received SUTENT were 65 years and older. No overall differences in safety or effectiveness were observed between these patients and younger patients.
In the pNET study, 22 (27%) of the patients who received SUTENT were 65 years and older. Clinical studies of SUTENT did not include sufficient numbers of patients with pNET to determine if patients 65 years of age and older respond differently than younger patients.
5.1 Hepatotoxicity
SUTENT can cause severe hepatotoxicity, resulting in liver failure or death. In the pooled safety population, liver failure occurred in <1% of patients in clinical trials. Liver failure include jaundiced, elevated transaminases and/or hyperbilirubinemia in conjunction with encephalopathy, coagulopathy, and/or renal failure.
Monitor liver function tests (alanine aminotransferase [ALT], aspartate aminotransferase [AST], and bilirubin) at baseline, during each cycle, and as clinically indicated. Interrupt SUTENT for Grade 3 hepatotoxicity until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose.
Discontinue SUTENT in patients with Grade 4 hepatotoxicity, in patients without resolution of Grade 3 hepatotoxicity, in patients who subsequently experience severe changes in liver function tests and in patients who have other signs and symptoms of liver failure. Safety in patients with ALT or AST >2.5 × upper limit of normal (ULN) or with >5 × ULN and liver metastases has not been established.
4 Contraindications (4 CONTRAINDICATIONS)
None.
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following clinically significant adverse reactions are described elsewhere in the labeling.
-
•Hepatotoxicity [see Warnings and Precautions (5.1)]
-
•Cardiovascular Events [see Warnings and Precautions (5.2)]
-
•QT Interval Prolongation and Torsade de Pointes [see Warnings and Precautions (5.3)]
-
•Hypertension [see Warnings and Precautions (5.4)]
-
•Hemorrhagic Events [see Warnings and Precautions (5.5)]
-
•Tumor Lysis Syndrome [see Warnings and Precautions (5.6)]
-
•Thrombotic Microangiopathy [see Warnings and Precautions (5.7)]
-
•Proteinuria [see Warnings and Precautions (5.8)]
-
•Dermatologic Toxicities [see Warnings and Precautions (5.9)]
-
•Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.10)]
-
•Thyroid Dysfunction [see Warnings and Precautions (5.11)]
-
•Hypoglycemia [see Warnings and Precautions (5.12)]
-
•Osteonecrosis of the Jaw [see Warnings and Precautions (5.13)]
-
•Impaired Wound Healing [see Warnings and Precautions (5.14)]
7 Drug Interactions (7 DRUG INTERACTIONS)
8.7 Renal Impairment
No dose adjustment is recommended in patients with mild (CLcr 50 to 80 mL/min), moderate (CLcr 30 to <50 mL/min), or severe (CLcr <30 mL/min) renal impairment who are not on dialysis [see Clinical Pharmacology (12.3)].
No dose adjustment is recommended for patients with end-stage renal disease (ESRD) on hemodialysis [see Clinical Pharmacology (12.3)].
12.3 Pharmacokinetics
The pharmacokinetics of sunitinib and sunitinib malate have been evaluated in healthy subjects and in patients with solid tumors.
Sunitinib AUC and Cmax increase proportionately over a dose range of 25 mg to 100 mg (0.5 to 2 times the approved RDD of 50 mg). The pharmacokinetics were similar in healthy subjects and in patients with a solid tumor, including patients with GIST and RCC. No significant changes in the pharmacokinetics of sunitinib or the primary active metabolite were observed with repeated daily administration or with repeated cycles. With repeated daily administration, sunitinib accumulates 3- to 4-fold while the primary metabolite accumulates 7- to 10-fold. Steady-state concentrations of sunitinib and its primary active metabolite are achieved within 10 to 14 days. By Day 14, combined plasma concentrations of sunitinib and its active metabolite ranged from 63 to 101 ng/mL.
8.6 Hepatic Impairment
No dose adjustment is required in patients with mild or moderate (Child-Pugh Class A or B) hepatic impairment [see Clinical Pharmacology (12.3)]. SUTENT was not studied in patients with severe (Child-Pugh Class C) hepatic impairment.
1 Indications and Usage (1 INDICATIONS AND USAGE)
SUTENT is a kinase inhibitor indicated for:
-
•treatment of adult patients with gastrointestinal stromal tumor (GIST) after disease progression on or intolerance to imatinib mesylate. (1.1)
-
•treatment of adult patients with advanced renal cell carcinoma (RCC). (1.2)
-
•adjuvant treatment of adult patients at high risk of recurrent RCC following nephrectomy. (1.3)
-
•treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNET) in adult patients with unresectable locally advanced or metastatic disease. (1.4)
Warning: Hepatotoxicity (WARNING: HEPATOTOXICITY)
Hepatotoxicity may be severe, and in some cases, fatal. Monitor hepatic function and interrupt, dose reduce, or discontinue SUTENT as recommended [see Warnings and Precautions (5.1)].
12.1 Mechanism of Action
Sunitinib is a small molecule that inhibits multiple receptor tyrosine kinases (RTKs), some of which are implicated in tumor growth, pathologic angiogenesis, and metastatic progression of cancer. Sunitinib was evaluated for its inhibitory activity against a variety of kinases (>80 kinases) and was identified as an inhibitor of platelet-derived growth factor receptors (PDGFRα and PDGFRβ), vascular endothelial growth factor receptors (VEGFR1, VEGFR2, and VEGFR3), stem cell factor receptor (KIT), Fms-like tyrosine kinase-3 (FLT3), colony stimulating factor receptor Type 1 (CSF-1R), and the glial cell-line derived neurotrophic factor receptor (RET). Sunitinib inhibition of the activity of these RTKs has been demonstrated in biochemical and cellular assays, and inhibition of function has been demonstrated in cell proliferation assays. The primary metabolite exhibits similar potency compared to sunitinib in biochemical and cellular assays.
Sunitinib inhibited the phosphorylation of multiple RTKs (PDGFRβ, VEGFR2, KIT) in tumor xenografts expressing RTK targets in vivo and demonstrated inhibition of tumor growth or tumor regression and/or inhibited metastases in some experimental models of cancer. Sunitinib demonstrated the ability to inhibit growth of tumor cells expressing dysregulated target RTKs (PDGFR, RET, or KIT) in vitro and to inhibit PDGFRβ- and VEGFR2-dependent tumor angiogenesis in vivo.
5.11 Thyroid Dysfunction
Hyperthyroidism, some followed by hypothyroidism, have been reported in clinical trials and through postmarketing experience of SUTENT.
Monitor thyroid function at baseline, periodically during treatment and as clinically indicated. Monitor patients closely for signs and symptoms of thyroid dysfunction, including hypothyroidism, hyperthyroidism, and thyroiditis, during treatment with SUTENT. Initiate and/or adjust therapies for thyroid dysfunction as appropriate.
13 Nonclinical Toxicology (13 NONCLINICAL TOXICOLOGY)
5.2 Cardiovascular Events
Cardiovascular events, including heart failure, cardiomyopathy, myocardial ischemia, and myocardial infarction, some of which were fatal, have been reported.
In pooled safety population, 3% of patients experienced heart failure; 71% of the patients with heart failure were reported as recovered. Fatal cardiac failure was reported in <1% of patients.
In the adjuvant treatment of RCC study, 11 patients experienced Grade 2 decreased ejection fraction (left ventricular ejection fraction [LVEF] 40% to 50% and a 10% to 19% decrease from baseline). In 3 of these 11 patients, the ejection fractions arm did not return to ≥50% or baseline by the time of last measurement. No patients who received SUTENT were diagnosed with CHF.
Patients who presented with cardiac events within 12 months prior to SUTENT administration, such as myocardial infarction (including severe/unstable angina), coronary/peripheral artery bypass graft, symptomatic CHF, cerebrovascular accident or transient ischemic attack, or pulmonary embolism were excluded from SUTENT clinical studies. Patients with prior anthracycline use or cardiac radiation were also excluded from some studies. It is unknown whether patients with these concomitant conditions may be at a higher risk of developing left ventricular dysfunction.
Consider monitoring LVEF at baseline and periodically as clinically indicated. Carefully monitor patients for clinical signs and symptoms of congestive heart failure (CHF). Discontinue SUTENT in patients who experience clinical manifestations of CHF. Interrupt SUTENT and/or reduce the dose in patients without clinical evidence of CHF who have an ejection fraction of greater than 20% but less than 50% below baseline or below the lower limit of normal if baseline ejection fraction was not obtained.
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
-
•Hepatotoxicity: Fatal liver failure has been observed. Monitor liver function tests at baseline, during each cycle, and as clinically indicated. Interrupt SUTENT for Grade 3 hepatotoxicity until resolution to Grade ≤1 or baseline and resume SUTENT at a reduced dose; discontinue if no resolution. Discontinue SUTENT in patients with Grade 4 hepatoxicity, in patients who have subsequent severe changes in liver function tests or other signs and symptoms of liver failure. (2.4, 5.1)
-
•Cardiovascular Events: Myocardial ischemia, myocardial infarction, heart failure, cardiomyopathy, and decreased left ventricular ejection fraction (LVEF) to below the lower limit of normal including death have occurred. Monitor for signs and symptoms of congestive heart failure and consider monitoring LVEF at baseline and periodically during treatment. Discontinue SUTENT for clinical manifestations of congestive heart failure. Interrupt and/or dose reduce for decreased LVEF. (5.2)
-
•QT Interval Prolongation and Torsade de Pointes: Monitor patients at higher risk for developing QT interval prolongation. Consider monitoring of electrocardiograms and electrolytes. (5.3)
-
•Hypertension: Monitor blood pressure at baseline and as clinically indicated. Initiate and/or adjust antihypertensive therapy as appropriate. Interrupt SUTENT for Grade 3 hypertension until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose. Discontinue SUTENT in patients who develop Grade 4 hypertension. (5.4)
-
•Hemorrhagic Events: Tumor-related hemorrhage and viscus perforation (both with fatal events) have occurred. Perform serial complete blood counts and physical examinations. Interrupt SUTENT for Grade 3 or 4 hemorrhagic events until resolution to Grade ≤1 or baseline, then resume at a reduced dose; discontinue if no resolution. (5.5)
-
•Tumor Lysis Syndrome (TLS): TLS (some fatal) has been reported primarily in patients with RCC and GIST. Monitor these patients and treat as clinically indicated. (5.6)
-
•Thrombotic microangiopathy (TMA): TMA, including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome, sometimes leading to renal failure or a fatal outcome, has been reported. Discontinue SUTENT for TMA. (5.7)
-
•Proteinuria: Renal failure or a fatal outcome has occurred. Monitor urine protein. Interrupt treatment for 24-hour urine protein of 3 or more grams. Discontinue for repeat episodes of 24-hour urine protein of 3 or more grams despite dose reductions or nephrotic syndrome. (5.8)
-
•Dermatologic Toxicities: Necrotizing fasciitis, erythema multiforme, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) (some fatal) have occurred. Discontinue SUTENT for these events. (5.9)
-
•Reversible Posterior Leukoencephalopathy Syndrome (RPLS): RPLS (some fatal) has been reported. Monitor for signs and symptoms of RPLS. Withhold SUTENT until resolution. (5.10)
-
•Thyroid Dysfunction: Monitor thyroid function at baseline, periodically during treatment, and as clinically indicated. Initiate and/or adjust therapy for thyroid dysfunction as appropriate. (5.11)
-
•Hypoglycemia: Check blood glucose levels regularly and assess if antidiabetic drug dose modifications are required. (5.12)
-
•Osteonecrosis of the Jaw (ONJ): Withhold SUTENT for at least 3 weeks prior to invasive dental procedure and for development of ONJ until complete resolution. (5.13)
-
•Impaired Wound Healing: Withhold SUTENT for at least 3 weeks prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of SUTENT after resolution of wound healing complications has not been established. (5.14)
-
•Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.15, 8.1, 8.3)
5.15 Embryo Fetal Toxicity (5.15 Embryo-Fetal Toxicity)
Based on findings from animal studies and its mechanism of action, SUTENT can cause fetal harm when administered to pregnant woman. Administration of sunitinib to pregnant rats and rabbits during the period of organogenesis resulted in teratogenicity at approximately 5.5 and 0.3 times the combined systemic exposure [combined area under the curve (AUC) of sunitinib plus its active metabolite] in patients administered the recommended daily dose (RDD) of 50 mg, respectively.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with SUTENT and for 4 weeks following the final dose [see Use in Specific Populations (8.1, 8.3)].
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
GIST and Advanced RCC:
-
•The recommended dosage is 50 mg orally once daily for the first 4 weeks of each 6-week cycle (Schedule 4/2). (2.1)
Adjuvant Treatment of RCC:
-
•The recommended dosage is 50 mg orally once daily for the first 4 weeks of a 6-week cycle (Schedule 4/2) for a maximum of 9 cycles. (2.2)
pNET:
-
•The recommended dosage is 37.5 mg orally once daily. (2.3)
5.14 Impaired Wound Healing
Impaired wound healing has been reported in patients who received SUTENT [see Adverse Reactions (6.2)].
Withhold SUTENT for at least 3 weeks prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of SUTENT after resolution of wound healing complications has not been established.
5.9 Dermatologic Toxicities
Severe cutaneous adverse reactions have been reported, including erythema multiforme (EM), Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN), some of which were fatal. Permanently discontinue SUTENT for these severe cutaneous adverse reactions.
Necrotizing fasciitis, including fatal cases, has been reported in patients treated with SUTENT, including of the perineum and secondary to fistula formation. Discontinue SUTENT in patients who develop necrotizing fasciitis.
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Capsules, hard gelatin:
-
•12.5 mg sunitinib: orange cap and orange body, printed with white ink "Pfizer" on the cap and "STN 12.5 mg" on the body.
-
•25 mg sunitinib: caramel cap and orange body, printed with white ink "Pfizer" on the cap and "STN 25 mg" on the body.
-
•37.5 mg sunitinib: yellow cap and yellow body, printed with black ink "Pfizer" on the cap and "STN 37.5 mg" on the body.
-
•50 mg sunitinib: caramel top and caramel body, printed with white ink "Pfizer" on the cap and "STN 50 mg" on the body.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of SUTENT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
•Blood and lymphatic system disorders: hemorrhage associated with thrombocytopeniaincluding some fatalities.
-
•Gastrointestinal disorders: esophagitis.
-
•Hepatobiliary disorders: cholecystitis, particularly acalculous cholecystitis.
-
•Immune system disorders: hypersensitivity reactions, including angioedema.
-
•Infections and infestations: serious infection (with or without neutropenia). The infections most commonly observed with SUTENT include respiratory, urinary tract, skin infections, and sepsis/septic shock.
-
•Musculoskeletal and connective tissue disorders: fistula formation, sometimes associated with tumor necrosis and/or regression; myopathy and/or rhabdomyolysis with or without acute renal failure.
-
•Renal and urinary disorders: renal impairment and/or failure.
-
•Respiratory disorders: pulmonary embolism, pleural effusion.
-
•Skin and subcutaneous tissue disorders: pyoderma gangrenosum, including positive de-challenges.
-
•Vascular disorders: arterial (including aortic) aneurysms, dissections, and rupture; arterial thromboembolic events. The most frequent events included cerebrovascular accident, transient ischemic attack, and cerebral infarction.
-
•General disorders and administration site conditions: impaired wound healing.
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
-
•Lactation: Advise not to breastfeed. (8.2)
5.6 Tumor Lysis Syndrome (tls) (5.6 Tumor Lysis Syndrome (TLS))
Tumor Lysis Syndrome (TLS), some fatal, occurred in clinical trials and has been reported in postmarketing experience, primarily in patients with RCC or GIST. Patients generally at risk of TLS are those with high tumor burden prior to treatment. Monitor these patients for TLS and manage as appropriate.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the Warnings and Precautions reflect exposure to SUTENT in 7527 patients with GIST, RCC (advanced and adjuvant), or pNET. In this pooled safety population, the most common adverse reactions (≥25%) were fatigue/asthenia, diarrhea, mucositis/stomatitis, nausea, decreased appetite/anorexia, vomiting, abdominal pain, hand-foot syndrome, hypertension, bleeding events, dysgeusia/altered taste, dyspepsia, and thrombocytopenia.
2.3 Recommended Dosage for Pnet (2.3 Recommended Dosage for pNET)
The recommended dosage of SUTENT for pancreatic neuroendocrine tumors (pNET) is 37.5 mg taken orally once daily until disease progression or unacceptable toxicity. SUTENT may be taken with or without food.
1.2 Advanced Renal Cell Carcinoma
SUTENT is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC).
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
1.1 Gastrointestinal Stromal Tumor
SUTENT is indicated for the treatment of adult patients with gastrointestinal stromal tumor (GIST) after disease progression on or intolerance to imatinib mesylate.
7.2 Drugs That Prolong Qt Interval (7.2 Drugs that Prolong QT Interval)
SUTENT is associated with QTc interval prolongation [see Warnings and Precautions (5.3), Clinical Pharmacology (12.2)]. Monitor the QT interval with ECGs more frequently in patients who require treatment with concomitant medications known to prolong the QT interval.
5.13 Osteonecrosis of the Jaw (onj) (5.13 Osteonecrosis of the Jaw (ONJ))
Osteonecrosis of the Jaw (ONJ) occurred in patients treated with SUTENT. Concomitant exposure to other risk factors, such as bisphosphonates or dental disease/invasive dental procedures, may increase the risk of ONJ. Perform an oral examination prior to initiation of SUTENT and periodically during SUTENT therapy. Advise patients regarding good oral hygiene practices. Withhold SUTENT treatment for at least 3 weeks prior to scheduled dental surgery or invasive dental procedures, if possible. Withhold SUTENT for development of ONJ until complete resolution. The safety of resumption of SUTENT after resolution of osteonecrosis of the jaw has not been established.
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
SUTENT 12.5 mg capsules are supplied as hard gelatin capsule with orange cap and orange body, printed with white ink "Pfizer" on the cap, "STN 12.5 mg" on the body:
Bottles of 28 capsules: NDC 0069-0550-38
SUTENT 25 mg capsules are supplied as hard gelatin capsule with caramel cap and orange body, printed with white ink "Pfizer" on the cap, "STN 25 mg" on the body:
Bottles of 28 capsules: NDC 0069-0770-38
SUTENT 37.5 mg capsules are supplied as hard gelatin capsule with yellow cap and yellow body, printed with black ink "Pfizer" on the cap, "STN 37.5 mg" on the body:
Bottles of 28 capsules: NDC 0069-0830-38
SUTENT 50 mg capsules are supplied as hard gelatin capsule with caramel cap and caramel body, printed with white ink "Pfizer" on the cap, "STN 50 mg" on the body:
Bottles of 28 capsules: NDC 0069-0980-38
5.7 Thrombotic Microangiopathy (tma) (5.7 Thrombotic Microangiopathy (TMA))
Thrombotic Microangiopathy (TMA), including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome, sometimes leading to renal failure or a fatal outcome, occurred in clinical trials and in postmarketing experience of SUTENT as monotherapy and administered in combination with bevacizumab. SUTENT is not approved for use in combination with bevacizumab.
Discontinue SUTENT in patients developing TMA. Reversal of the effects of TMA has been observed after SUTENT was discontinued.
14.3 Pancreatic Neuroendocrine Tumors
Study 6 (NCT#00428597) was a multi-center, international, randomized, double-blind, placebo-controlled study of single-agent SUTENT conducted in patients with unresectable pNET. Patients were required to have documented RECIST-defined disease progression within the prior 12 months and were randomized (1:1) to receive either 37.5 mg SUTENT (N=86) or placebo (N=85) once daily without a scheduled off-treatment period. The primary objective was to compare PFS in patients receiving SUTENT versus patients receiving placebo. Other endpoints included OS, ORR, and safety. Use of somatostatin analogs was allowed in the study.
Demographics were comparable between the SUTENT and placebo groups. Additionally, 49% of SUTENT patients had nonfunctioning tumors vs 52% of placebo patients, and 92% patients in both arms had liver metastases. A total of 66% of SUTENT patients received prior systemic therapy compared with 72% of placebo patients and 35% of SUTENT patients had received somatostatin analogs compared with 38% of placebo patients. Patients were treated until disease progression or withdrawal from the study. Upon disease progression or study closure, patients were offered access to SUTENT in a separate extension study.
As recommended by the Independent Data Monitoring Committee, the study was terminated prematurely prior to the prespecified interim analysis. This may have led to an overestimate of the magnitude of PFS effect. A clinically significant improvement for SUTENT over placebo in PFS was seen by both investigator and independent assessment. A hazard ratio favoring SUTENT was observed in all subgroups of baseline characteristics evaluated. OS data were not mature at the time of the analysis. There were 9 deaths in the SUTENT arm and 21 deaths in the placebo arm. A statistically significant difference in ORR favoring SUTENT over placebo was observed. Efficacy results are summarized in Table 15 and the Kaplan-Meier curve for PFS is in Figure 4.
| Abbreviations: CI=confidence interval; HR=hazard ratio; N=number of patients; NA=not applicable; pNET=pancreatic neuroendocrine tumors. | ||||
|
Efficacy Parameter |
SUTENT
|
Placebo
|
p-value |
HR
|
|
Progression-free survival |
10.2 |
5.4 |
0.000146 2-sided unstratified log-rank test.
|
0.427 |
|
Objective response rate |
9.3 |
0 |
0.0066 Fisher's Exact test.
|
NA |
Figure 4. Kaplan-Meier Curve of PFS in the pNET Study 6
Abbreviations: CI=confidence interval; N=number of patients; PFS=progression-free survival; pNET=pancreatic neuroendocrine tumors.
1.4 Advanced Pancreatic Neuroendocrine Tumors
SUTENT is indicated for the treatment of progressive, well-differentiated pancreatic neuroendocrine tumors (pNET) in adult patients with unresectable locally advanced or metastatic disease.
5.5 Hemorrhagic Events and Viscus Perforation
Hemorrhagic events, some of which were fatal, have involved the gastrointestinal tract, respiratory tract, tumor, urinary tract, and brain. In the pooled safety population, 30% of patients experienced hemorrhagic events, including Grade 3 or 4 in 4.2% of patients. Epistaxis was the most common hemorrhagic event and gastrointestinal hemorrhage was the most common Grade 3–5 event.
Tumor-related hemorrhage was observed in patients treated with SUTENT. These events may occur suddenly, and in the case of pulmonary tumors, may present as severe and life-threatening hemoptysis or pulmonary hemorrhage. Pulmonary hemorrhage, some with a fatal outcome, was observed in patients treated with SUTENT for metastatic RCC, GIST, and metastatic lung cancer. SUTENT is not approved for use in patients with lung cancer.
Serious, sometimes fatal, gastrointestinal complications including gastrointestinal perforation, have been reported in patients with intra-abdominal malignancies treated with SUTENT.
Include serial complete blood counts (CBCs) and physical examinations with the clinical assessment of hemorrhagic events. Interrupt SUTENT for Grade 3 or 4 hemorrhagic events until resolution to Grade ≤1 or baseline, then resume SUTENT at a reduced dose.
Discontinue SUTENT in patients without resolution of Grade 3 or 4 hemorrhagic events.
1.3 Adjuvant Treatment of Renal Cell Carcinoma
SUTENT is indicated for the adjuvant treatment of adult patients at high risk of recurrent RCC following nephrectomy.
2.4 Dosage Modifications for Adverse Reactions
To manage adverse reactions, the recommended dosage modifications are provided in Table 1. Table 2 provides the recommended dosage reductions of SUTENT for adverse reactions.
|
Indications |
GIST |
RCC |
pNET |
|
|
Advanced RCC |
Adjuvant RCC |
|||
|
First dose reduction |
37.5 mg once daily |
37.5 mg once daily |
37.5 mg once daily |
25 mg once daily |
|
Second dose reduction |
25 mg once daily |
25 mg once daily |
NA |
NA |
|
Adverse Reaction |
Severity |
Dosage Modifications for SUTENT |
|
Hepatotoxicity [see Warnings and Precautions (5.1)] |
Grade 3 |
|
|
Grade 4 |
|
|
|
Cardiovascular events [see Warnings and Precautions (5.2)] |
Asymptomatic cardiomyopathy (left ventricular ejection fraction greater than 20% but less than 50% below baseline or below the lower limit of normal if baseline was not obtained) |
|
|
Clinically manifested congestive heart failure (CHF) |
|
|
|
Hypertension [see Warnings and Precautions (5.4)] |
Grade 3 |
|
|
Grade 4 |
|
|
|
Hemorrhagic events [see Warnings and Precautions (5.5)] |
Grade 3 or 4 |
|
|
Thrombotic microangiopathy [see Warnings and Precautions (5.7)] |
Any Grade |
|
|
Proteinuria or Nephrotic syndrome [see Warnings and Precautions (5.8)] |
3 or more grams proteinuria in 24 hours in the absence of nephrotic syndrome |
|
|
Nephrotic syndrome or recurrent proteinuria of 3 or more grams per 24 hours despite dose reductions |
|
|
|
Dermatological toxicities Erythema multiforme (EM), Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), Necrotizing fasciitis [see Warnings and Precautions (5.9)] |
Any Grade |
|
|
Reversible posterior leukoencephalopathy syndrome [see Warnings and Precautions (5.10)] |
Any Grade |
|
|
Osteonecrosis of the jaw [see Warnings and Precautions (5.13)] |
Any Grade |
|
|
Impaired wound healing [see Warnings and Precautions (5.14)] |
Any Grade |
|
8.3 Females and Males of Reproductive Potential
SUTENT can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
2.1 Recommended Dosage for Gist and Advanced Rcc (2.1 Recommended Dosage for GIST and Advanced RCC)
The recommended dosage of SUTENT for gastrointestinal stromal tumor (GIST) and advanced renal cell carcinoma (RCC) is 50 mg taken orally once daily, on a schedule of 4 weeks on treatment followed by 2 weeks off (Schedule 4/2) until disease progression or unacceptable toxicity. SUTENT may be taken with or without food.
5.3 Qt Interval Prolongation and Torsade De Pointes (5.3 QT Interval Prolongation and Torsade de Pointes)
SUTENT can cause QT interval prolongation in a dose-dependent manner, which may lead to an increased risk for ventricular arrhythmias including Torsade de Pointes. Torsade de Pointes was observed in <0.1% of patients.
Monitor patients who are at higher risk of developing QT interval prolongation, including patients with a history of QT interval prolongation, patients who are taking antiarrhythmics, or patients with relevant pre-existing cardiac disease, bradycardia, or electrolyte disturbances. Consider periodic monitoring of electrocardiograms and electrolytes (i.e., magnesium, potassium) during treatment with SUTENT.
Monitor QT interval more frequently when SUTENT is concomitantly administered with strong CYP3A4 inhibitors or drugs known to prolong QT interval. Consider dose reducing SUTENT [see Dosage and Administration (2.5), Drug Interactions (7.2)].
2.2 Recommended Dosage for Adjuvant Treatment of Rcc (2.2 Recommended Dosage for Adjuvant Treatment of RCC)
The recommended dosage of SUTENT for the adjuvant treatment of RCC is 50 mg taken orally once daily, on a schedule of 4 weeks on treatment followed by 2 weeks off (Schedule 4/2), for nine 6-week cycles. SUTENT may be taken with or without food.
Principal Display Panel 50 Mg Capsule Bottle Label (PRINCIPAL DISPLAY PANEL - 50 mg Capsule Bottle Label)
PROFESSIONAL SAMPLE – NOT FOR SALE
ALWAYS DISPENSE WITH MEDICATION GUIDE
Pfizer
NDC 63539-019-01
Sutent®
(sunitinib malate)
50 mg*
Capsules
14 Capsules
Rx only
Principal Display Panel 12.5 Mg Capsule Bottle Label (PRINCIPAL DISPLAY PANEL - 12.5 mg Capsule Bottle Label)
PROFESSIONAL SAMPLE – NOT FOR SALE
ALWAYS DISPENSE WITH MEDICATION GUIDE
Pfizer
NDC 63539-017-01
Sutent®
(sunitinib malate)
12.5 mg*
Capsules
28 Capsules
Rx only
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of sunitinib has been evaluated in 2 species: rasH2 transgenic mice and Sprague-Dawley rats. There were similar positive findings in both species. In rasH2 transgenic mice, gastroduodenal carcinomas and/or gastric mucosal hyperplasia, as well as an increased incidence of background hemangiosarcomas were observed at sunitinib daily doses of ≥25 mg/kg/day in studies of 1 or 6 months duration. No proliferative changes were observed in rasH2 transgenic mice at 8 mg/kg/day. Similarly, in a 2-year rat carcinogenicity study, administration of sunitinib in 28-day cycles followed by 7-day dose-free periods resulted in findings of duodenal carcinoma at doses as low as 1 mg/kg/day [approximately 0.9 times the combined AUC (combined systemic exposure of sunitinib plus its active metabolite) in patients administered the RDD of 50 mg]. At the high dose of 3 mg/kg/day (approximately 8 times the combined AUC in patients administered the RDD of 50 mg), the incidence of duodenal tumors was increased and was accompanied by findings of gastric mucous cell hyperplasia and by an increased incidence of pheochromocytoma and hyperplasia of the adrenal gland.
Sunitinib did not cause genetic damage when tested in in vitro assays [bacterial mutation (Ames test), human lymphocyte chromosome aberration] and an in vivo rat bone marrow micronucleus test.
In a female fertility and early embryonic development study, female rats were administered oral sunitinib (0.5, 1.5, 5 mg/kg/day) for 21 days prior to mating and for 7 days after mating. Preimplantation loss was observed in females administered 5 mg/kg/day (approximately 5 times the combined AUC in patients administered the RDD of 50 mg). No adverse effects on fertility were observed at doses ≤1.5 mg/kg/day (approximately equal to the combined AUC in patients administered the RDD of 50 mg). In addition, effects on the female reproductive system were identified in a 3-month oral repeat-dose monkey study (2, 6, 12 mg/kg/day). Ovarian changes (decreased follicular development) were noted at 12 mg/kg/day (approximately 5 times the combined AUC in patients administered the RDD of 50 mg), while uterine changes (endometrial atrophy) were noted at ≥2 mg/kg/day (approximately 0.4 times the combined AUC in patients administered the RDD of 50 mg). With the addition of vaginal atrophy, the uterine and ovarian effects were reproduced at 6 mg/kg/day (approximately 0.8 times the combined AUC in patients administered the RDD of 50 mg) in a 9-month monkey study (0.3, 1.5, and 6 mg/kg/day administered daily for 28 days followed by a 14-day respite).
In a male fertility study, no reproductive effects were observed in male rats dosed with 1, 3, or 10 mg/kg/day oral sunitinib for 58 days prior to mating with untreated females. Fertility, copulation, conception indices, and sperm evaluation (morphology, concentration, and motility) were unaffected by sunitinib at doses ≤10 mg/kg/day (approximately ≥26 times the combined AUC in patients administered the RDD of 50 mg).
5.10 Reversible Posterior Leukoencephalopathy Syndrome (rpls) (5.10 Reversible Posterior Leukoencephalopathy Syndrome (RPLS))
Reversible posterior leukoencephalopathy syndrome (RPLS) has been reported in <1% of patients, some of which were fatal. Patients can present with hypertension, headache, decreased alertness, altered mental functioning, and visual loss, including cortical blindness. Magnetic resonance imaging is necessary to confirm the diagnosis. Discontinue SUTENT in patients developing RPLS.
2.6 Dosage Modification for End Stage Renal Disease Patients On Hemodialysis (2.6 Dosage Modification for End-Stage Renal Disease Patients on Hemodialysis)
No starting dose adjustment is required in patients with end-stage renal disease (ESRD) on hemodialysis. However, given the decreased exposure compared to patients with normal renal function, subsequent doses may be increased gradually up to 2-fold based on safety and tolerability [see Clinical Pharmacology (12.3)].
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:51:22.901241 · Updated: 2026-03-14T22:40:12.922880