Amjevita
9c44bd5b-4464-40a4-8933-8680950f00f2
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
AMJEVITA is a tumor necrosis factor (TNF) blocker indicated for: Reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis . ( 1.1 ) Reducing signs and symptoms of moderately to severely active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older. ( 1.2 ) Reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with active psoriatic arthritis . ( 1.3 ) Reducing signs and symptoms in adult patients with active ankylosing spondylitis . ( 1.4 ) Treatment of moderately to severely active Crohn's disease in adults and pediatric patients 6 years of age and older. ( 1.5 ) Treatment of moderately to severely active ulcerative colitis in adult patients. ( 1.6 ) Limitations of Use: Effectiveness has not been established in patients who have lost response to or were intolerant to TNF blockers. Treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy, and when other systemic therapies are medically less appropriate. ( 1.7 ) Treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older. ( 1.8 ) Treatment of non-infectious intermediate, posterior, and panuveitis in adults and pediatric patients 2 years of age and older. ( 1.9 )
Dosage and Administration
Administer by subcutaneous injection ( 2 ) Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis ( 2.2 ): Adults: 40 mg every other week. Some patients with RA not receiving methotrexate may benefit from increasing the dosage to 40 mg every week or 80 mg every other week. Juvenile Idiopathic Arthritis or Pediatric Uveitis ( 2.3 ): Pediatric Weight 2 Years of Age and Older Recommended Dosage 10 kg (22 lbs) to less than 15 kg (33 lbs) 10 mg every other week 15 kg (33 lbs) to less than 30 kg (66 lbs) 20 mg every other week 30 kg (66 lbs) and greater 40 mg every other week Crohn's Disease ( 2.4 ): Adults: 160 mg on Day 1 (given in one day or split over two consecutive days); 80 mg on Day 15; and 40 mg every other week starting on Day 29. Pediatric Patients 6 Years of Age and Older: Pediatric Weight Recommended Dosage Days 1 and 15 Starting on Day 29 17 kg (37 lbs) to less than 40 kg (88 lbs) Day 1: 80 mg Day 15: 40 mg 20 mg every other week 40 kg (88 lbs) and greater Day 1: 160 mg (single-dose or split over two consecutive days) Day 15: 80 mg 40 mg every other week Ulcerative Colitis ( 2.5 ): Adults: 160 mg on Day 1 (given in one day or split over two consecutive days), 80 mg on Day 15 and 40 mg every other week starting on Day 29. Discontinue in patients without evidence of clinical remission by eight weeks (Day 57). Plaque Psoriasis or Adult Uveitis ( 2.6 ): Adults: 80 mg initial dose, followed by 40 mg every other week starting one week after initial dose. Hidradenitis Suppurativa ( 2.7 ) Adults: Day 1:160 mg (given in one day or split over two consecutive days) Day 15: 80 mg Day 29 and subsequent doses: 40 mg every week or 80 mg every other week Adolescents 12 years of age and older: Adolescent Weight Recommended Dosage 30 kg (66 lbs) to less than 60 kg (132 lbs) Day 1: 80 mg Day 8 and subsequent doses: 40 mg every other week 60 kg (132 lbs) and greater Day 1: 160 mg (given in one day or split over two consecutive days) Day 15: 80 mg Day 29 and subsequent doses: 40 mg every week or 80 mg every other week
Contraindications
None.
Warnings and Precautions
Serious infections: Do not start AMJEVITA during an active infection. If an infection develops, monitor carefully, and stop AMJEVITA if infection becomes serious. ( 5.1 ) Invasive fungal infections: For patients who develop a systemic illness on AMJEVITA, consider empiric antifungal therapy for those who reside or travel to regions where mycoses are endemic. ( 5.1 ) Malignancies: Incidence of malignancies was greater in adalimumab-treated patients than in controls. ( 5.2 ) Anaphylaxis or serious hypersensitivity reactions may occur. ( 5.3 ) Hepatitis B virus reactivation: Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop AMJEVITA and begin anti-viral therapy. ( 5.4 ) Demyelinating disease: Exacerbation or new onset, may occur. ( 5.5 ) Cytopenias, pancytopenia: Advise patients to seek immediate medical attention if symptoms develop, and consider stopping AMJEVITA. ( 5.6 ) Heart failure: Worsening or new onset, may occur. ( 5.8 ) Autoimmunity: Stop AMJEVITA if lupus-like syndrome or autoimmune hepatitis develop. ( 5.9 )
Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling: Serious Infections [see Warnings and Precautions (5.1) ] Malignancies [see Warnings and Precautions (5.2) ] Hypersensitivity Reactions [see Warnings and Precautions (5.3) ] Hepatitis B Virus Reactivation [see Warnings and Precautions (5.4) ] Neurologic Reactions [see Warnings and Precautions (5.5) ] Hematological Reactions [see Warnings and Precautions (5.6) ] Heart Failure [see Warnings and Precautions (5.8) ] Autoimmunity [see Warnings and Precautions (5.9) ]
Drug Interactions
Abatacept: Increased risk of serious infection. ( 5.1 , 5.11 , 7.2 ) Anakinra: Increased risk of serious infection. ( 5.1 , 5.7 , 7.2 ) Live vaccines: Avoid use with AMJEVITA. ( 5.10 , 7.3 )
How Supplied
AMJEVITA ® (adalimumab-atto) injection is supplied as a preservative-free, sterile, clear, colorless to slightly yellow solution for subcutaneous administration. AMJEVITA is supplied in a single-dose prefilled syringe (PFS) or single-dose prefilled SureClick autoinjector (AI). The AMJEVITA prefilled syringe and prefilled SureClick autoinjector are not made with natural rubber latex. The following packaging configurations are available. Presentation Number of Units/Pack NDC number 10 mg/0.2 mL prefilled glass syringe with a fixed 29 gauge needle 1 55513-413-01 20 mg/0.2 mL prefilled syringe with a fixed 29 gauge needle 1 55513-399-01 72511-399-01 20 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle 1 55513-411-01 40 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle 1 55513-479-01 2 55513-479-02 72511-479-02 40 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle 1 55513-410-01 2 55513-410-02 80 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle 1 55513-480-01 2 55513-480-02 40 mg/0.4 mL Prefilled SureClick Autoinjector 1 55513-482-01 72511-482-01 2 55513-482-02 72511-482-02 40 mg/0.8 mL Prefilled SureClick Autoinjector 1 55513-400-01 72511-400-01 2 55513-400-02 72511-400-02 80 mg/0.8 mL Prefilled SureClick Autoinjector 1 55513-481-01 2 55513-481-02 72511-481-02
Storage and Handling
AMJEVITA ® (adalimumab-atto) injection is supplied as a preservative-free, sterile, clear, colorless to slightly yellow solution for subcutaneous administration. AMJEVITA is supplied in a single-dose prefilled syringe (PFS) or single-dose prefilled SureClick autoinjector (AI). The AMJEVITA prefilled syringe and prefilled SureClick autoinjector are not made with natural rubber latex. The following packaging configurations are available. Presentation Number of Units/Pack NDC number 10 mg/0.2 mL prefilled glass syringe with a fixed 29 gauge needle 1 55513-413-01 20 mg/0.2 mL prefilled syringe with a fixed 29 gauge needle 1 55513-399-01 72511-399-01 20 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle 1 55513-411-01 40 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle 1 55513-479-01 2 55513-479-02 72511-479-02 40 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle 1 55513-410-01 2 55513-410-02 80 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle 1 55513-480-01 2 55513-480-02 40 mg/0.4 mL Prefilled SureClick Autoinjector 1 55513-482-01 72511-482-01 2 55513-482-02 72511-482-02 40 mg/0.8 mL Prefilled SureClick Autoinjector 1 55513-400-01 72511-400-01 2 55513-400-02 72511-400-02 80 mg/0.8 mL Prefilled SureClick Autoinjector 1 55513-481-01 2 55513-481-02 72511-481-02
Description
WARNING: SERIOUS INFECTIONS AND MALIGNANCY See full prescribing information for complete boxed warning. SERIOUS INFECTIONS ( 5.1 , 6.1 ): Increased risk of serious infections leading to hospitalization or death, including tuberculosis (TB), bacterial sepsis, invasive fungal infections (such as histoplasmosis), and infections due to other opportunistic pathogens. Discontinue AMJEVITA if a patient develops a serious infection or sepsis during treatment. Perform test for latent TB; if positive, start treatment for TB prior to starting AMJEVITA. Monitor all patients for active TB during treatment, even if initial latent TB test is negative. MALIGNANCY ( 5.2 ): Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers including adalimumab products. Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have occurred in adolescent and young adults with inflammatory bowel disease treated with TNF blockers including adalimumab products.
Medication Information
Warnings and Precautions
Serious infections: Do not start AMJEVITA during an active infection. If an infection develops, monitor carefully, and stop AMJEVITA if infection becomes serious. ( 5.1 ) Invasive fungal infections: For patients who develop a systemic illness on AMJEVITA, consider empiric antifungal therapy for those who reside or travel to regions where mycoses are endemic. ( 5.1 ) Malignancies: Incidence of malignancies was greater in adalimumab-treated patients than in controls. ( 5.2 ) Anaphylaxis or serious hypersensitivity reactions may occur. ( 5.3 ) Hepatitis B virus reactivation: Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop AMJEVITA and begin anti-viral therapy. ( 5.4 ) Demyelinating disease: Exacerbation or new onset, may occur. ( 5.5 ) Cytopenias, pancytopenia: Advise patients to seek immediate medical attention if symptoms develop, and consider stopping AMJEVITA. ( 5.6 ) Heart failure: Worsening or new onset, may occur. ( 5.8 ) Autoimmunity: Stop AMJEVITA if lupus-like syndrome or autoimmune hepatitis develop. ( 5.9 )
Indications and Usage
AMJEVITA is a tumor necrosis factor (TNF) blocker indicated for: Reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis . ( 1.1 ) Reducing signs and symptoms of moderately to severely active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older. ( 1.2 ) Reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with active psoriatic arthritis . ( 1.3 ) Reducing signs and symptoms in adult patients with active ankylosing spondylitis . ( 1.4 ) Treatment of moderately to severely active Crohn's disease in adults and pediatric patients 6 years of age and older. ( 1.5 ) Treatment of moderately to severely active ulcerative colitis in adult patients. ( 1.6 ) Limitations of Use: Effectiveness has not been established in patients who have lost response to or were intolerant to TNF blockers. Treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy, and when other systemic therapies are medically less appropriate. ( 1.7 ) Treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older. ( 1.8 ) Treatment of non-infectious intermediate, posterior, and panuveitis in adults and pediatric patients 2 years of age and older. ( 1.9 )
Dosage and Administration
Administer by subcutaneous injection ( 2 ) Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis ( 2.2 ): Adults: 40 mg every other week. Some patients with RA not receiving methotrexate may benefit from increasing the dosage to 40 mg every week or 80 mg every other week. Juvenile Idiopathic Arthritis or Pediatric Uveitis ( 2.3 ): Pediatric Weight 2 Years of Age and Older Recommended Dosage 10 kg (22 lbs) to less than 15 kg (33 lbs) 10 mg every other week 15 kg (33 lbs) to less than 30 kg (66 lbs) 20 mg every other week 30 kg (66 lbs) and greater 40 mg every other week Crohn's Disease ( 2.4 ): Adults: 160 mg on Day 1 (given in one day or split over two consecutive days); 80 mg on Day 15; and 40 mg every other week starting on Day 29. Pediatric Patients 6 Years of Age and Older: Pediatric Weight Recommended Dosage Days 1 and 15 Starting on Day 29 17 kg (37 lbs) to less than 40 kg (88 lbs) Day 1: 80 mg Day 15: 40 mg 20 mg every other week 40 kg (88 lbs) and greater Day 1: 160 mg (single-dose or split over two consecutive days) Day 15: 80 mg 40 mg every other week Ulcerative Colitis ( 2.5 ): Adults: 160 mg on Day 1 (given in one day or split over two consecutive days), 80 mg on Day 15 and 40 mg every other week starting on Day 29. Discontinue in patients without evidence of clinical remission by eight weeks (Day 57). Plaque Psoriasis or Adult Uveitis ( 2.6 ): Adults: 80 mg initial dose, followed by 40 mg every other week starting one week after initial dose. Hidradenitis Suppurativa ( 2.7 ) Adults: Day 1:160 mg (given in one day or split over two consecutive days) Day 15: 80 mg Day 29 and subsequent doses: 40 mg every week or 80 mg every other week Adolescents 12 years of age and older: Adolescent Weight Recommended Dosage 30 kg (66 lbs) to less than 60 kg (132 lbs) Day 1: 80 mg Day 8 and subsequent doses: 40 mg every other week 60 kg (132 lbs) and greater Day 1: 160 mg (given in one day or split over two consecutive days) Day 15: 80 mg Day 29 and subsequent doses: 40 mg every week or 80 mg every other week
Contraindications
None.
Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling: Serious Infections [see Warnings and Precautions (5.1) ] Malignancies [see Warnings and Precautions (5.2) ] Hypersensitivity Reactions [see Warnings and Precautions (5.3) ] Hepatitis B Virus Reactivation [see Warnings and Precautions (5.4) ] Neurologic Reactions [see Warnings and Precautions (5.5) ] Hematological Reactions [see Warnings and Precautions (5.6) ] Heart Failure [see Warnings and Precautions (5.8) ] Autoimmunity [see Warnings and Precautions (5.9) ]
Drug Interactions
Abatacept: Increased risk of serious infection. ( 5.1 , 5.11 , 7.2 ) Anakinra: Increased risk of serious infection. ( 5.1 , 5.7 , 7.2 ) Live vaccines: Avoid use with AMJEVITA. ( 5.10 , 7.3 )
Storage and Handling
AMJEVITA ® (adalimumab-atto) injection is supplied as a preservative-free, sterile, clear, colorless to slightly yellow solution for subcutaneous administration. AMJEVITA is supplied in a single-dose prefilled syringe (PFS) or single-dose prefilled SureClick autoinjector (AI). The AMJEVITA prefilled syringe and prefilled SureClick autoinjector are not made with natural rubber latex. The following packaging configurations are available. Presentation Number of Units/Pack NDC number 10 mg/0.2 mL prefilled glass syringe with a fixed 29 gauge needle 1 55513-413-01 20 mg/0.2 mL prefilled syringe with a fixed 29 gauge needle 1 55513-399-01 72511-399-01 20 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle 1 55513-411-01 40 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle 1 55513-479-01 2 55513-479-02 72511-479-02 40 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle 1 55513-410-01 2 55513-410-02 80 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle 1 55513-480-01 2 55513-480-02 40 mg/0.4 mL Prefilled SureClick Autoinjector 1 55513-482-01 72511-482-01 2 55513-482-02 72511-482-02 40 mg/0.8 mL Prefilled SureClick Autoinjector 1 55513-400-01 72511-400-01 2 55513-400-02 72511-400-02 80 mg/0.8 mL Prefilled SureClick Autoinjector 1 55513-481-01 2 55513-481-02 72511-481-02
How Supplied
AMJEVITA ® (adalimumab-atto) injection is supplied as a preservative-free, sterile, clear, colorless to slightly yellow solution for subcutaneous administration. AMJEVITA is supplied in a single-dose prefilled syringe (PFS) or single-dose prefilled SureClick autoinjector (AI). The AMJEVITA prefilled syringe and prefilled SureClick autoinjector are not made with natural rubber latex. The following packaging configurations are available. Presentation Number of Units/Pack NDC number 10 mg/0.2 mL prefilled glass syringe with a fixed 29 gauge needle 1 55513-413-01 20 mg/0.2 mL prefilled syringe with a fixed 29 gauge needle 1 55513-399-01 72511-399-01 20 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle 1 55513-411-01 40 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle 1 55513-479-01 2 55513-479-02 72511-479-02 40 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle 1 55513-410-01 2 55513-410-02 80 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle 1 55513-480-01 2 55513-480-02 40 mg/0.4 mL Prefilled SureClick Autoinjector 1 55513-482-01 72511-482-01 2 55513-482-02 72511-482-02 40 mg/0.8 mL Prefilled SureClick Autoinjector 1 55513-400-01 72511-400-01 2 55513-400-02 72511-400-02 80 mg/0.8 mL Prefilled SureClick Autoinjector 1 55513-481-01 2 55513-481-02 72511-481-02
Description
WARNING: SERIOUS INFECTIONS AND MALIGNANCY See full prescribing information for complete boxed warning. SERIOUS INFECTIONS ( 5.1 , 6.1 ): Increased risk of serious infections leading to hospitalization or death, including tuberculosis (TB), bacterial sepsis, invasive fungal infections (such as histoplasmosis), and infections due to other opportunistic pathogens. Discontinue AMJEVITA if a patient develops a serious infection or sepsis during treatment. Perform test for latent TB; if positive, start treatment for TB prior to starting AMJEVITA. Monitor all patients for active TB during treatment, even if initial latent TB test is negative. MALIGNANCY ( 5.2 ): Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers including adalimumab products. Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have occurred in adolescent and young adults with inflammatory bowel disease treated with TNF blockers including adalimumab products.
Section 42229-5
SERIOUS INFECTIONS
Patients treated with adalimumab products, including AMJEVITA, are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
Discontinue AMJEVITA if a patient develops a serious infection or sepsis.
Reported infections include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients with TB have frequently presented with disseminated or extrapulmonary disease. Test patients for latent TB before AMJEVITA use and during therapy. Initiate treatment for latent TB prior to AMJEVITA use.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. Consider empiric anti-fungal therapy in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral and other infections due to opportunistic pathogens, including Legionella and Listeria.
Carefully consider the risks and benefits of treatment with AMJEVITA prior to initiating therapy in patients with chronic or recurrent infection.
Monitor patients closely for the development of signs and symptoms of infection during and after treatment with AMJEVITA, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Section 42231-1
| MEDICATION GUIDE AMJEVITA® (am-jeh-vee'-tah) (adalimumab-atto) injection, for subcutaneous use |
||
|---|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 10/2025 | |
| Read the Medication Guide that comes with AMJEVITA before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment. | ||
|
What is the most important information I should know about AMJEVITA?
Before starting AMJEVITA, tell your doctor if you:
|
||
|
|
|
AMJEVITA can make you more likely to get infections or make any infection that you may have worse. Cancer
|
||
|
What is AMJEVITA?
AMJEVITA is a medicine called a Tumor Necrosis Factor (TNF) blocker. AMJEVITA is used:
|
||
|
What should I tell my doctor before taking AMJEVITA?
AMJEVITA may not be right for you. Before starting AMJEVITA, tell your doctor about all of your medical conditions, including if you:
Especially tell your doctor if you use:
|
||
How should I take AMJEVITA?
|
||
|
What are the possible side effects of AMJEVITA?
AMJEVITA can cause serious side effects, including: See "What is the most important information I should know about AMJEVITA?"
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
The most common side effects of AMJEVITA include:
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store AMJEVITA?
|
||
|
General information about the safe and effective use of AMJEVITA
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use AMJEVITA for a condition for which it was not prescribed. Do not give AMJEVITA to other people, even if they have the same condition. It may harm them. This Medication Guide summarizes the most important information about AMJEVITA. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about AMJEVITA that is written for health professionals. |
||
|
What are the ingredients in AMJEVITA?
Active ingredient: adalimumab-atto Inactive ingredients: glacial acetic acid (or L-lactic acid), polysorbate 80, sucrose and Water for Injection. Sodium hydroxide is added as necessary to adjust the pH to 5.2. AMJEVITA® (adalimumab-atto) Manufactured by: Amgen Inc., One Amgen Center Drive, Thousand Oaks, CA 91320-1799 U.S.A U.S. License Number 1080 © 2022–2025 Amgen Inc. All rights reserved.1xxxxxx – v10 For more information call 1-800-77-AMGEN (1-800-772-6436). |
Section 43683-2
Section 44425-7
Storage and Stability
Do not use beyond the expiration date on the container. AMJEVITA must be refrigerated at 36°F to 46°F (2°C to 8°C). DO NOT FREEZE. Do not use if frozen even if it has been thawed.
Store in original carton until time of administration to protect from light.
If needed, for example when traveling, AMJEVITA may be stored at room temperature up to a maximum of 77°F (25°C) for a period of up to 14 days, with protection from light. AMJEVITA should be discarded if not used within the 14-day period. Record the date when AMJEVITA is first removed from the refrigerator in the spaces provided on the carton.
Do not store AMJEVITA in extreme heat or cold.
1.9 Uveitis
AMJEVITA is indicated for the treatment of non-infectious intermediate, posterior, and panuveitis in adults and pediatric patients 2 years of age and older.
15 References
- National Cancer Institute. Surveillance, Epidemiology, and End Results Database (SEER) Program. SEER Incidence Crude Rates, 17 Registries, 2000-2007.
10. Overdosage
Doses up to 10 mg/kg have been administered to patients in clinical trials without evidence of dose-limiting toxicities. In case of overdosage, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions or effects and appropriate symptomatic treatment instituted immediately.
Consider contacting the Poison Help line (1-800-222-1222) or medical toxicologist for additional overdose management recommendations.
11 Description
Adalimumab-atto is a tumor necrosis factor blocker. Adalimumab-atto is a recombinant human IgG1 monoclonal antibody with human derived heavy and light chain variable regions and human IgG1:k constant regions. Adalimumab-atto is produced by recombinant DNA technology in a mammalian cell (Chinese Hamster Ovary (CHO)) expression system and is purified by a process that includes specific viral inactivation and removal steps. It consists of 1330 amino acids and has a molecular weight of approximately 148 kilodaltons.
AMJEVITA® (adalimumab-atto) injection is supplied as a sterile, preservative-free solution for subcutaneous administration. The drug product is supplied as either a single-dose, prefilled SureClick autoinjector, or as a single-dose, 1 mL prefilled glass syringe. Enclosed within the autoinjector is a single-dose, 1 mL prefilled glass syringe. The solution of AMJEVITA is clear, colorless to slightly yellow, with a pH of about 5.2.
Each 80 mg/0.8 mL prefilled syringe or prefilled autoinjector delivers 0.8 mL (80 mg) of drug product. Each 0.8 mL of AMJEVITA is formulated with L-lactic acid (1.7 mg), polysorbate 80 (0.8 mg), sodium hydroxide for pH adjustment, sucrose (67 mg), and Water for Injection, USP, pH 5.2.
Each 40 mg/0.8 mL prefilled syringe or prefilled autoinjector delivers 0.8 mL (40 mg) of drug product. Each 0.8 mL of AMJEVITA is formulated with glacial acetic acid (0.48 mg), polysorbate 80 (0.8 mg), sodium hydroxide for pH adjustment, sucrose (72 mg), and Water for Injection, USP, pH 5.2.
Each 40 mg/0.4 mL prefilled syringe or prefilled autoinjector delivers 0.4 mL (40 mg) of drug product. Each 0.4 mL of AMJEVITA is formulated with L-lactic acid (0.9 mg), polysorbate 80 (0.4 mg), sodium hydroxide for pH adjustment, sucrose (34 mg), and Water for Injection, USP, pH 5.2.
Each 20 mg/0.4 mL prefilled syringe delivers 0.4 mL (20 mg) of drug product. Each 0.4 mL of AMJEVITA is formulated with glacial acetic acid (0.24 mg), polysorbate 80 (0.4 mg), sodium hydroxide for pH adjustment, sucrose (36 mg), and Water for Injection, USP, pH 5.2.
Each 20 mg/0.2 mL prefilled syringe delivers 0.2 mL (20 mg) of drug product. Each 0.2 mL of AMJEVITA is formulated with L-lactic acid (0.4 mg), polysorbate 80 (0.2 mg), sodium hydroxide for pH adjustment, sucrose (17 mg), and Water for Injection, USP, pH 5.2.
Each 10 mg/0.2 mL prefilled syringe delivers 0.2 mL (10 mg) of drug product. Each 0.2 mL of AMJEVITA is formulated with glacial acetic acid (0.12 mg), polysorbate 80 (0.2 mg), sodium hydroxide for pH adjustment, sucrose (18 mg), and Water for Injection, USP, pH 5.2.
5.2 Malignancies
Consider the risks and benefits of TNF-blocker treatment including AMJEVITA prior to initiating therapy in patients with a known malignancy other than a successfully treated non-melanoma skin cancer (NMSC) or when considering continuing a TNF blocker in patients who develop a malignancy.
5.9 Autoimmunity
Treatment with adalimumab products may result in the formation of autoantibodies and, rarely, in the development of a lupus-like syndrome or autoimmune hepatitis [see Adverse Reactions (6.1, 6.3)]. If a patient develops symptoms and findings suggestive of a lupus-like syndrome or autoimmune hepatitis following treatment with AMJEVITA, discontinue treatment and evaluate the patient.
7.1 Methotrexate
Adalimumab has been studied in rheumatoid arthritis (RA) patients taking concomitant methotrexate (MTX). Although MTX reduced the apparent clearance of adalimumab, the data do not suggest the need for dose adjustment of either AMJEVITA or MTX [see Clinical Pharmacology (12.3)].
5.8 Heart Failure
Cases of worsening congestive heart failure (CHF) and new onset CHF have been reported with TNF blockers. Cases of worsening CHF have also been observed with adalimumab products. Adalimumab products have not been formally studied in patients with CHF; however, in clinical trials of another TNF blocker, a higher rate of serious CHF-related adverse reactions was observed. Exercise caution when using AMJEVITA in patients who have heart failure and monitor them carefully.
7.3 Live Vaccines
Avoid the use of live vaccines with AMJEVITA [see Warnings and Precautions (5.10)].
8.4 Pediatric Use
The safety and effectiveness of AMJEVITA have not been established in pediatric patients with psoriatic arthritis, ankylosing spondylitis, or plaque psoriasis.
The safety and effectiveness of AMJEVITA have been established for:
- reducing signs and symptoms of moderately to severely active polyarticular JIA in pediatric patients 2 years of age and older.
- the treatment of moderately to severely active Crohn's disease in pediatric patients 6 years of age and older.
- the treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older.
- the treatment of non-infectious intermediate, posterior, and panuveitis in pediatric patients 2 years of age and older.
A pediatric assessment for AMJEVITA demonstrates that AMJEVITA is safe and effective for pediatric patients in an indication for which HUMIRA (adalimumab) is approved. However, AMJEVITA is not approved for such indication due to marketing exclusivity for HUMIRA (adalimumab).
Due to their inhibition of TNFα, adalimumab products administered during pregnancy could affect immune response in the in utero-exposed newborn and infant. Data from eight infants exposed to adalimumab in utero suggest adalimumab crosses the placenta [see Use in Specific Populations (8.1)]. The clinical significance of elevated adalimumab concentrations in infants is unknown. The safety of administering live or live-attenuated vaccines in exposed infants is unknown. Risks and benefits should be considered prior to vaccinating (live or live-attenuated) exposed infants.
Post-marketing cases of lymphoma, including hepatosplenic T-cell lymphoma and other malignancies, some fatal, have been reported among children, adolescents, and young adults who received treatment with TNF-blockers including adalimumab products [see Warnings and Precautions (5.2)].
8.5 Geriatric Use
In clinical studies of RA (Studies RA-I, RA-II, RA-III, and RA-IV), a total of 519 subjects 65 years of age and older, including 107 subjects 75 years of age and older, received adalimumab. No overall difference in effectiveness was observed between these subjects and younger adult subjects.
The frequency of serious infection and malignancy among adalimumab-treated subjects 65 years of age and older was higher than for those less than 65 years of age. Consider the benefits and risks of AMJEVITA in patients 65 years of age and older. In patients treated with AMJEVITA, closely monitor for the development of infection or malignancy [see Warnings and Precautions (5.1, 5.2)].
5.10 Immunizations
In a placebo-controlled clinical trial of patients with RA, no difference was detected in anti-pneumococcal antibody response between adalimumab and placebo treatment groups when the pneumococcal polysaccharide vaccine and influenza vaccine were administered concurrently with adalimumab. Similar proportions of subjects developed protective levels of anti-influenza antibodies between adalimumab and placebo treatment groups; however, titers in aggregate to influenza antigens were moderately lower in patients receiving adalimumab. The clinical significance of this is unknown. Patients on AMJEVITA may receive concurrent vaccinations, except for live vaccines. No data are available on the secondary transmission of infection by live vaccines in patients receiving adalimumab products.
It is recommended that pediatric patients, if possible, be brought up to date with all immunizations in agreement with current immunization guidelines prior to initiating AMJEVITA therapy. Patients on AMJEVITA may receive concurrent vaccinations, except for live vaccines.
The safety of administering live or live-attenuated vaccines in infants exposed to adalimumab products in utero is unknown. Risks and benefits should be considered prior to vaccinating (live or live-attenuated) exposed infants [see Use in Specific Populations (8.1 , 8.4)].
6.2 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of adalimumab or of other adalimumab products.
There are two assays that have been used to measure anti-adalimumab antibodies. With the ELISA, antibodies to adalimumab could be detected only when serum adalimumab concentrations were < 2 mcg/mL. The ECL assay can detect anti-adalimumab antibody titers independent of adalimumab concentrations in the serum samples. The incidence of anti-adalimumab antibody (AAA) development in patients treated with adalimumab are presented in Table 2.
| Indications | Study Duration | Anti-Adalimumab Antibody Incidence by ELISA (n/N) | Anti-Adalimumab Antibody Incidence by ECL Assay (n/N) | ||
|---|---|---|---|---|---|
| In all patients who received adalimumab | In patients with serum adalimumab concentrations < 2 mcg/mL | ||||
| n: number of patients with anti-adalimumab antibody; NR: not reported; NA: Not applicable (not performed) | |||||
| Rheumatoid Arthritis In patients receiving concomitant methotrexate (MTX), the incidence of anti-adalimumab antibody was 1% compared to 12% with adalimumab monotherapy
|
6 to 12 months | 5% (58/1062) | NR | NA | |
| Juvenile Idiopathic Arthritis (JIA) | 4 to 17 years of age In patients receiving concomitant MTX, the incidence of anti-adalimumab antibody was 6% compared to 26% with adalimumab monotherapy
|
48 weeks | 16% (27/171) | NR | NA |
| 2 to 4 years of age or ≥ 4 years of age and weighing < 15 kg | 24 weeks | 7% (1/15) This patient received concomitant MTX
|
NR | NA | |
| Psoriatic Arthritis In patients receiving concomitant MTX, the incidence of antibody development was 7% compared to 1% in RA
|
48 weeks Subjects enrolled after completing 2 previous studies of 24 weeks or 12 weeks of treatments
|
13% (24/178) | NR | NA | |
| Ankylosing Spondylitis | 24 weeks | 9% (16/185) | NR | NA | |
| Adult Crohn's Disease | 56 weeks | 3% (7/269) | 8% (7/86) | NA | |
| Pediatric Crohn's Disease | 52 weeks | 3% (6/182) | 10% (6/58) | NA | |
| Adult Ulcerative Colitis | 52 weeks | 5% (19/360) | 21% (19/92) | NA | |
| Plaque Psoriasis In plaque psoriasis patients who were on adalimumab monotherapy and subsequently withdrawn from the treatment, the rate of antibodies to adalimumab after retreatment was similar to the rate observed prior to withdrawal
|
Up to 52 weeks One 12-week Phase 2 study and one 52-week Phase 3 study
|
8% (77/920) | 21% (77/372) | NA | |
| Hidradenitis Suppurativa | 36 weeks | 7% (30/461) | 28% (58/207) Among subjects in the two Phase 3 studies who stopped adalimumab treatment for up to 24 weeks and in whom adalimumab serum levels subsequently declined to < 2 mcg/mL (approximately 22% of total subjects studied)
|
61% (272/445 No apparent association between antibody development and safety was observed
|
|
| Non-infectious Uveitis | 52 weeks | 5% (12/249) | 21% (12/57) | 40% (99/249) No correlation of antibody development to safety or efficacy outcomes was observed
|
1.5 Crohn's Disease
AMJEVITA is indicated for the treatment of moderately to severely active Crohn's disease in adults and pediatric patients 6 years of age and older.
4 Contraindications
None.
6 Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Malignancies [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Hepatitis B Virus Reactivation [see Warnings and Precautions (5.4)]
- Neurologic Reactions [see Warnings and Precautions (5.5)]
- Hematological Reactions [see Warnings and Precautions (5.6)]
- Heart Failure [see Warnings and Precautions (5.8)]
- Autoimmunity [see Warnings and Precautions (5.9)]
7 Drug Interactions
1.7 Plaque Psoriasis
AMJEVITA is indicated for the treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy, and when other systemic therapies are medically less appropriate. AMJEVITA should only be administered to patients who will be closely monitored and have regular follow-up visits with a physician [see Warnings and Precautions (5)].
Instructions for Use
AMJEVITA® (am-jeh-vee'-tah)
(adalimumab-atto)
injection, for subcutaneous use
40 mg/0.8 mL
single-dose prefilled SureClick® autoinjector
This Instructions for Use contains information on how to inject AMJEVITA with a SureClick autoinjector.
If your healthcare provider decides that you or a caregiver may be able to give your injections of AMJEVITA at home, you should receive training on the right way to prepare and inject AMJEVITA. Do not try to inject yourself until you have been shown the right way to give the injections by your healthcare provider or nurse.
The medicine in the AMJEVITA autoinjector is for injection under the skin (subcutaneous injection). See the AMJEVITA Medication Guide for information about AMJEVITA.
| Getting to know your prefilled autoinjector |
1. Important Information You Need to Know Before Injecting AMJEVITA
- It is important that you do not try to give the injection until you have fully read and understood this Instructions for Use.
- Do not use the autoinjector if the carton is damaged or the seal is broken.
- Do not use the autoinjector after the expiration date on the label.
- Do not shake the autoinjector.
- Do not remove the yellow cap from the autoinjector until you are ready to inject.
- Do not use the autoinjector if it has been frozen.
- Do not use the autoinjector if it has been dropped on a hard surface. Part of the autoinjector may be broken even if you cannot see the break. Use a new autoinjector, and call 1-800-77-AMGEN (1-800-772-6436).
- The autoinjector is not made with natural rubber latex.
| Important: Keep the autoinjector and sharps disposal container out of the sight and reach of children. |
Frequently asked questions:
For additional information and answers to frequently asked questions, visit www.amjevita.com.
Where to get help:
If you want more information or help using AMJEVITA:
- Contact your healthcare provider,
- Visit www.amjevita.com, or
- Call 1-800-77-AMGEN (1-800-772-6436)
2. Storing and Preparing to Inject AMJEVITA
2a Refrigerate the autoinjector carton until you are ready to use it.
- Keep the autoinjector in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Keep the autoinjector in the original carton to protect it from light or physical damage.
- Do not freeze the autoinjector.
- Do not store the autoinjector in extreme heat or cold. For example, avoid storing in your vehicle's glove box or trunk.
| Important: Keep the autoinjector out of the sight and reach of children. |
| WAIT |
2b Wait 15 to 30 minutes for the autoinjector to reach room temperature.
- Remove the number of autoinjectors you need for your injection and put any unused autoinjectors back into the refrigerator.
- Let the autoinjector warm up naturally.
- Do not heat the autoinjector with hot water, a microwave, or direct sunlight.
- Do not shake the autoinjector at any time.
- Using the autoinjector at room temperature makes sure the full dose is delivered and allows for a more comfortable injection.
2c You may keep AMJEVITA at room temperature for up to 14 days, if needed.
- For example, when you are traveling, you may keep AMJEVITA at room temperature.
- Keep it at room temperature between 68°F to 77°F (20°C to 25°C).
- Do not put it back in the refrigerator.
- Record the date you removed it from the refrigerator and use it within 14 days.
| Important: Place the autoinjector in a sharps disposal container if it has reached room temperature and has not been used within 14 days. |
2d Inspect the medicine. It should be clear and colorless to pale yellow.
- It is okay to see air bubbles in the autoinjector.
- Do not use AMJEVITA if the medicine is cloudy, discolored, or has flakes or particles.
| Important: If the medicine is cloudy, discolored, or has flakes or particles, or if the autoinjector is damaged or expired, call 1-800-77-AMGEN (1-800-772-6436). |
2e Check the expiration date (Exp.) and inspect the autoinjector for damage.
- Do not use the autoinjector if the expiration date has passed.
-
Do not use the autoinjector if:
- the yellow cap is missing or loose.
- it has cracks or broken parts.
- it has been dropped on a hard surface.
- Make sure you have the right medicine and dose.
3. Getting Ready for Your Injection
3a Gather and place the following items for your injection on a clean, flat, and well-lit surface:
- AMJEVITA autoinjector (room temperature)
- Sharps disposal container [see Disposing of AMJEVITA and Checking the Injection Site]
- Alcohol wipe
- Adhesive bandage
- Cotton ball or gauze pad
3b Inject into 1 of these locations.
- Inject into the front of your thigh or stomach (except for 2 inches around your belly button).
- Choose a different site for each injection.
| Important: Avoid areas with scars or stretch marks, or where the skin is tender, bruised, red, or hard. |
3c Wash hands thoroughly with soap and water.
3d Clean the injection site with an alcohol wipe.
- Let the skin dry on its own.
- Do not touch this area again before injecting.
4. Injecting AMJEVITA
| Important: Only remove the yellow cap when you can inject right away (within 5 minutes) because the medicine can dry out. Do not recap. |
4a Grasp the autoinjector so you can see the window. Pull the yellow cap straight off. You may need to pull hard.
- Do not twist, bend or wiggle the yellow cap to pull it off.
- Never put the yellow cap back on. It may damage the needle.
- Do not put your finger inside the cream safety guard.
- It is normal to see a drop of medicine at the end of the needle or cream safety guard.
4b Pinch the skin to create a firm surface at the injection site. Place the cream safety guard straight against the pinched skin.
- Keep the skin pinched until the injection is finished.
- Make sure you can see the window.
- Make sure the autoinjector is positioned straight on the injection site (at a 90-degree angle).
|
PUSH and hold against skin |
4c Firmly push the autoinjector down until the cream safety guard stops moving. Hold the autoinjector down, do not lift.
- The cream safety guard pushes in and unlocks the light blue start button.
|
PRESS light blue start button |
4d Keep pushing the autoinjector down and press the light blue start button to start the injection.
- You may hear or feel a click.
- The window starts to turn yellow.
- It is okay to let go of the light blue start button.
|
WATCH and CONFIRM that the window turns fully yellow |
4e Keep pushing the autoinjector down. When the window is fully yellow, the injection is complete.
- The injection may take up to 10 seconds to complete.
- You may hear or feel a click.
- Lift the autoinjector away from your skin.
- The cream safety guard locks around the needle.
| Important: If the window has not turned fully yellow, or it looks like the medicine is still coming out, you have not received a full dose. Call your healthcare provider right away. |
5. Disposing of AMJEVITA and Checking the Injection Site
5a Place the used autoinjector and yellow cap in an FDA-cleared sharps disposal container right away after use.
| Important: Do not throw away the autoinjector in your household trash. |
- Do not reuse the autoinjector.
- Do not touch the cream safety guard.
5b Check the injection site.
- Do not rub the injection site.
- If there is blood, press a cotton ball or gauze pad on your injection site. Apply an adhesive bandage if necessary.
Additional information about your sharps disposal container
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
Disposing of sharps disposal containers:
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container.
There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at:
http://www.fda.gov/safesharpsdisposal
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
Do not recycle your used sharps disposal container.
For more information or help call 1-800-77-AMGEN (1-800-772-6436).
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
AMJEVITA (adalimumab-atto)
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
US License Number 1080
©2024 Amgen Inc. All rights reserved.
1xxxxxx
Revised: 8/2024
v2
12.2 Pharmacodynamics
After treatment with adalimumab, a decrease in concentrations of acute phase reactants of inflammation (C-reactive protein [CRP] and erythrocyte sedimentation rate [ESR]) and serum cytokines (IL-6) was observed compared to baseline in patients with rheumatoid arthritis. A decrease in CRP concentrations was also observed in patients with Crohn's disease, ulcerative colitis, and hidradenitis suppurativa. Serum concentrations of matrix metalloproteinases (MMP-1 and MMP-3) that produce tissue remodeling responsible for cartilage destruction were also decreased after adalimumab administration.
12.3 Pharmacokinetics
The pharmacokinetics of adalimumab were linear over the dose range of 0.5 to 10 mg/kg following administration of a single intravenous dose (adalimumab products are not approved for intravenous use). Following 20, 40, and 80 mg every other week and every week subcutaneous administration, adalimumab mean serum trough concentrations at steady state increased approximately proportionally with dose in RA patients. The mean terminal half-life was approximately 2 weeks, ranging from 10 to 20 days across studies. Healthy subjects and patients with RA displayed similar adalimumab pharmacokinetics.
Adalimumab exposure in patients treated with 80 mg every other week is estimated to be comparable with that in patients treated with 40 mg every week.
1.6 Ulcerative Colitis
AMJEVITA is indicated for the treatment of moderately to severely active ulcerative colitis in adult patients.
5.1 Serious Infections
Patients treated with adalimumab products, including AMJEVITA, are at increased risk for developing serious infections involving various organ systems and sites that may lead to hospitalization or death. Opportunistic infections due to bacterial, mycobacterial, invasive fungal, viral, parasitic, or other opportunistic pathogens including aspergillosis, blastomycosis, candidiasis, coccidioidomycosis, histoplasmosis, legionellosis, listeriosis, pneumocystosis and tuberculosis have been reported with TNF blockers. Patients have frequently presented with disseminated rather than localized disease.
The concomitant use of a TNF blocker and abatacept or anakinra was associated with a higher risk of serious infections in patients with rheumatoid arthritis (RA); therefore, the concomitant use of AMJEVITA and these biologic products is not recommended in the treatment of patients with RA [see Warnings and Precautions (5.7, 5.11) and Drug Interactions (7.2)].
Treatment with AMJEVITA should not be initiated in patients with an active infection, including localized infections. Patients 65 years of age and older, patients with co-morbid conditions and/or patients taking concomitant immunosuppressants (such as corticosteroids or methotrexate), may be at greater risk of infection. Consider the risks and benefits of treatment prior to initiating therapy in patients:
- with chronic or recurrent infection;
- who have been exposed to tuberculosis;
- with a history of an opportunistic infection;
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses, such as histoplasmosis, coccidioidomycosis, or blastomycosis; or
- with underlying conditions that may predispose them to infection.
1 Indications and Usage
AMJEVITA is a tumor necrosis factor (TNF) blocker indicated for:
- Reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis. (1.1)
- Reducing signs and symptoms of moderately to severely active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older. (1.2)
- Reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with active psoriatic arthritis. (1.3)
- Reducing signs and symptoms in adult patients with active ankylosing spondylitis. (1.4)
- Treatment of moderately to severely active Crohn's disease in adults and pediatric patients 6 years of age and older. (1.5)
- Treatment of moderately to severely active ulcerative colitis in adult patients. (1.6)
Limitations of Use: Effectiveness has not been established in patients who have lost response to or were intolerant to TNF blockers. - Treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy, and when other systemic therapies are medically less appropriate. (1.7)
- Treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older. (1.8)
- Treatment of non-infectious intermediate, posterior, and panuveitis in adults and pediatric patients 2 years of age and older. (1.9)
1.3 Psoriatic Arthritis
AMJEVITA is indicated for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with active psoriatic arthritis.
AMJEVITA can be used alone or in combination with non-biologic DMARDs.
7.2 Biological Products
In clinical studies in patients with RA, an increased risk of serious infections has been observed with the combination of TNF blockers with anakinra or abatacept, with no added benefit; therefore, use of AMJEVITA with abatacept or anakinra is not recommended in patients with RA [see Warnings and Precautions (5.7, 5.11)]. A higher rate of serious infections has also been observed in patients with RA treated with rituximab who received subsequent treatment with a TNF blocker. There is insufficient information regarding the concomitant use of AMJEVITA and other biologic products for the treatment of RA, PsA, AS, CD, UC, Ps, HS and UV. Concomitant administration of AMJEVITA with other biologic DMARDs (e.g., anakinra and abatacept) or other TNF blockers is not recommended based upon the possible increased risk for infections and other potential pharmacological interactions.
1.1 Rheumatoid Arthritis
AMJEVITA is indicated for reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis. AMJEVITA can be used alone or in combination with methotrexate or other non-biologic disease-modifying anti-rheumatic drugs (DMARDs).
12.1 Mechanism of Action
Adalimumab products bind specifically to TNF-alpha and block its interaction with the p55 and p75 cell surface TNF receptors. Adalimumab products also lyse surface TNF expressing cells in vitro in the presence of complement. Adalimumab products do not bind or inactivate lymphotoxin (TNF-beta). TNF is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Elevated concentrations of TNF are found in the synovial fluid of patients with RA, JIA, PsA, and AS and play an important role in both the pathologic inflammation and the joint destruction that are hallmarks of these diseases. Increased concentrations of TNF are also found in psoriasis plaques. In Ps, treatment with AMJEVITA may reduce the epidermal thickness and infiltration of inflammatory cells. The relationship between these pharmacodynamic activities and the mechanism(s) by which adalimumab products exert their clinical effects is unknown.
Adalimumab products also modulate biological responses that are induced or regulated by TNF, including changes in the concentrations of adhesion molecules responsible for leukocyte migration (ELAM-1, VCAM-1, and ICAM-1 with an IC50 of 1-2 × 10-10M).
5.5 Neurologic Reactions
Use of TNF blocking agents, including adalimumab products, has been associated with rare cases of new onset or exacerbation of clinical symptoms and/or radiographic evidence of central nervous system demyelinating disease, including multiple sclerosis (MS) and optic neuritis, and peripheral demyelinating disease, including Guillain-Barré syndrome. Exercise caution in considering the use of AMJEVITA in patients with preexisting or recent-onset central or peripheral nervous system demyelinating disorders; discontinuation of AMJEVITA should be considered if any of these disorders develop. There is a known association between intermediate uveitis and central demyelinating disorders.
1.4 Ankylosing Spondylitis
AMJEVITA is indicated for reducing signs and symptoms in adult patients with active ankylosing spondylitis.
5 Warnings and Precautions
- Serious infections: Do not start AMJEVITA during an active infection. If an infection develops, monitor carefully, and stop AMJEVITA if infection becomes serious. (5.1)
- Invasive fungal infections: For patients who develop a systemic illness on AMJEVITA, consider empiric antifungal therapy for those who reside or travel to regions where mycoses are endemic. (5.1)
- Malignancies: Incidence of malignancies was greater in adalimumab-treated patients than in controls. (5.2)
- Anaphylaxis or serious hypersensitivity reactions may occur. (5.3)
- Hepatitis B virus reactivation: Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop AMJEVITA and begin anti-viral therapy. (5.4)
- Demyelinating disease: Exacerbation or new onset, may occur. (5.5)
- Cytopenias, pancytopenia: Advise patients to seek immediate medical attention if symptoms develop, and consider stopping AMJEVITA. (5.6)
- Heart failure: Worsening or new onset, may occur. (5.8)
- Autoimmunity: Stop AMJEVITA if lupus-like syndrome or autoimmune hepatitis develop. (5.9)
2 Dosage and Administration
- Administer by subcutaneous injection (2)
Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis (2.2): - Adults: 40 mg every other week.
Some patients with RA not receiving methotrexate may benefit from increasing the dosage to 40 mg every week or 80 mg every other week.
Juvenile Idiopathic Arthritis or Pediatric Uveitis (2.3):
| Pediatric Weight 2 Years of Age and Older |
Recommended Dosage |
|---|---|
| 10 kg (22 lbs) to less than 15 kg (33 lbs) | 10 mg every other week |
| 15 kg (33 lbs) to less than 30 kg (66 lbs) | 20 mg every other week |
| 30 kg (66 lbs) and greater | 40 mg every other week |
Crohn's Disease (2.4):
- Adults: 160 mg on Day 1 (given in one day or split over two consecutive days); 80 mg on Day 15; and 40 mg every other week starting on Day 29.
-
Pediatric Patients 6 Years of Age and Older:
Pediatric Weight Recommended Dosage Days 1 and 15 Starting on Day 29 17 kg (37 lbs) to less than 40 kg (88 lbs) Day 1: 80 mg
Day 15: 40 mg20 mg every other week 40 kg (88 lbs) and greater Day 1: 160 mg (single-dose or split over two consecutive days)
Day 15: 80 mg40 mg every other week
Ulcerative Colitis (2.5):
- Adults: 160 mg on Day 1 (given in one day or split over two consecutive days), 80 mg on Day 15 and 40 mg every other week starting on Day 29. Discontinue in patients without evidence of clinical remission by eight weeks (Day 57).
Plaque Psoriasis or Adult Uveitis (2.6):
- Adults: 80 mg initial dose, followed by 40 mg every other week starting one week after initial dose.
Hidradenitis Suppurativa (2.7)
-
Adults:
- Day 1:160 mg (given in one day or split over two consecutive days)
- Day 15: 80 mg
- Day 29 and subsequent doses: 40 mg every week or 80 mg every other week
-
Adolescents 12 years of age and older:
Adolescent Weight Recommended Dosage 30 kg (66 lbs) to less than 60 kg (132 lbs) Day 1: 80 mg
Day 8 and subsequent doses: 40 mg every other week60 kg (132 lbs) and greater Day 1: 160 mg (given in one day or split over two consecutive days)
Day 15: 80 mg
Day 29 and subsequent doses: 40 mg every week or 80 mg every other week
5.6 Hematological Reactions
Rare reports of pancytopenia including aplastic anemia have been reported with TNF blocking agents. Adverse reactions of the hematologic system, including medically significant cytopenia (e.g., thrombocytopenia, leukopenia) have been infrequently reported with adalimumab products. The causal relationship of these reports to adalimumab products remains unclear. Advise all patients to seek immediate medical attention if they develop signs and symptoms suggestive of blood dyscrasias or infection (e.g., persistent fever, bruising, bleeding, pallor) while on AMJEVITA. Consider discontinuation of AMJEVITA therapy in patients with confirmed significant hematologic abnormalities.
1.8 Hidradenitis Suppurativa
AMJEVITA is indicated for the treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older.
3 Dosage Forms and Strengths
AMJEVITA is a clear, colorless to slightly yellow solution available as:
-
Prefilled SureClick Autoinjector
-
Injection: 80 mg/0.8 mL in a single-dose prefilled SureClick autoinjector.
-
Injection: 40 mg/0.8 mL in a single-dose prefilled SureClick autoinjector.
-
Injection: 40 mg/0.4 mL in a single-dose prefilled SureClick autoinjector.
-
-
Prefilled Syringe
-
Injection: 80 mg/0.8 mL in a single-dose prefilled glass syringe.
-
Injection: 40 mg/0.8 mL in a single-dose prefilled glass syringe.
-
Injection: 40 mg/0.4 mL in a single-dose prefilled glass syringe.
-
Injection: 20 mg/0.4 mL in a single-dose prefilled glass syringe.
-
Injection: 20 mg/0.2 mL in a single-dose prefilled glass syringe.
-
Injection: 10 mg/0.2 mL in a single-dose prefilled glass syringe.
-
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of adalimumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to adalimumab products exposure.
Gastrointestinal disorders: Diverticulitis, large bowel perforations including perforations associated with diverticulitis and appendiceal perforations associated with appendicitis, pancreatitis
General disorders and administration site conditions: Pyrexia
Hepato-biliary disorders: Liver failure, hepatitis, autoimmune hepatitis
Immune system disorders: Sarcoidosis
Neoplasms benign, malignant and unspecified (including cysts and polyps): Merkel Cell Carcinoma (neuroendocrine carcinoma of the skin)
Nervous system disorders: Demyelinating disorders (e.g., optic neuritis, Guillain-Barré syndrome), cerebrovascular accident
Respiratory disorders: Interstitial lung disease, including pulmonary fibrosis, pulmonary embolism
Skin reactions: Stevens Johnson Syndrome, cutaneous vasculitis, erythema multiforme, new or worsening psoriasis (all sub-types including pustular and palmoplantar), alopecia, lichenoid skin reaction
Vascular disorders: Systemic vasculitis, deep vein thrombosis
5.3 Hypersensitivity Reactions
Anaphylaxis and angioneurotic edema have been reported following administration of adalimumab products. If an anaphylactic or other serious allergic reaction occurs, immediately discontinue administration of AMJEVITA and institute appropriate therapy. In clinical trials of adalimumab, hypersensitivity reactions (e.g., rash, anaphylactoid reaction, fixed drug reaction, non-specified drug reaction, urticaria) have been observed.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reaction with adalimumab was injection site reactions. In placebo-controlled trials, 20% of subjects treated with adalimumab developed injection site reactions (erythema and/or itching, hemorrhage, pain or swelling), compared to 14% of subjects receiving placebo. Most injection site reactions were described as mild and generally did not necessitate drug discontinuation.
The proportion of subjects who discontinued treatment due to adverse reactions during the double-blind, placebo-controlled portion of studies in subjects with RA (i.e., Studies RA-I, RA-II, RA-III and RA-IV) was 7% for subjects taking adalimumab and 4% for placebo-treated subjects. The most common adverse reactions leading to discontinuation of adalimumab in these RA studies were clinical flare reaction (0.7%), rash (0.3%) and pneumonia (0.3%).
7.4 Cytochrome P450 Substrates
The formation of CYP450 enzymes may be suppressed by increased concentrations of cytokines (e.g., TNFα, IL-6) during chronic inflammation. It is possible for products that antagonize cytokine activity, such as adalimumab products, to influence the formation of CYP450 enzymes. Upon initiation or discontinuation of AMJEVITA in patients being treated with CYP450 substrates with a narrow therapeutic index, monitoring of the effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) is recommended and the individual dose of the drug product may be adjusted as needed.
1.2 Juvenile Idiopathic Arthritis
AMJEVITA is indicated for reducing signs and symptoms of moderately to severely active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older. AMJEVITA can be used alone or in combination with methotrexate.
17 Patient Counseling Information
Advise the patient or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
5.4 Hepatitis B Virus Reactivation
Use of TNF blockers, including AMJEVITA, may increase the risk of reactivation of hepatitis B virus (HBV) in patients who are chronic carriers of this virus. In some instances, HBV reactivation occurring in conjunction with TNF blocker therapy has been fatal. The majority of these reports have occurred in patients concomitantly receiving other medications that suppress the immune system, which may also contribute to HBV reactivation. Evaluate patients at risk for HBV infection for prior evidence of HBV infection before initiating TNF blocker therapy. Exercise caution in prescribing TNF blockers for patients identified as carriers of HBV. Adequate data are not available on the safety or efficacy of treating patients who are carriers of HBV with anti-viral therapy in conjunction with TNF blocker therapy to prevent HBV reactivation. For patients who are carriers of HBV and require treatment with TNF blockers, closely monitor such patients for clinical and laboratory signs of active HBV infection throughout therapy and for several months following termination of therapy. In patients who develop HBV reactivation, stop AMJEVITA and initiate effective anti-viral therapy with appropriate supportive treatment. The safety of resuming TNF blocker therapy after HBV reactivation is controlled is not known. Therefore, exercise caution when considering resumption of AMJEVITA therapy in this situation and monitor patients closely.
16 How Supplied/storage and Handling
AMJEVITA® (adalimumab-atto) injection is supplied as a preservative-free, sterile, clear, colorless to slightly yellow solution for subcutaneous administration. AMJEVITA is supplied in a single-dose prefilled syringe (PFS) or single-dose prefilled SureClick autoinjector (AI). The AMJEVITA prefilled syringe and prefilled SureClick autoinjector are not made with natural rubber latex.
The following packaging configurations are available.
| Presentation | Number of Units/Pack | NDC number |
|---|---|---|
| 10 mg/0.2 mL prefilled glass syringe with a fixed 29 gauge needle | 1 | 55513-413-01 |
| 20 mg/0.2 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-399-01 |
| 72511-399-01 | ||
| 20 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-411-01 |
| 40 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-479-01 |
| 2 | 55513-479-02 | |
| 72511-479-02 | ||
| 40 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-410-01 |
| 2 | 55513-410-02 | |
| 80 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-480-01 |
| 2 | 55513-480-02 | |
| 40 mg/0.4 mL Prefilled SureClick Autoinjector | 1 | 55513-482-01 72511-482-01 |
| 2 | 55513-482-02 72511-482-02 |
|
| 40 mg/0.8 mL Prefilled SureClick Autoinjector | 1 | 55513-400-01 72511-400-01 |
| 2 | 55513-400-02 72511-400-02 |
|
| 80 mg/0.8 mL Prefilled SureClick Autoinjector | 1 | 55513-481-01 |
| 2 | 55513-481-02 | |
| 72511-481-02 |
2.1 Recommended Tuberculosis Evaluation
Prior to initiating AMJEVITA and periodically during therapy, evaluate patients for active tuberculosis and test for latent infection [see Warnings and Precautions (5.1)].
14.9 Clinical Studies in Plaque Psoriasis
The safety and efficacy of adalimumab were assessed in randomized, double-blind, placebo-controlled studies in 1696 adult subjects with moderate to severe chronic plaque psoriasis (Ps) who were candidates for systemic therapy or phototherapy.
Study Ps-I evaluated 1212 subjects with chronic Ps with ≥ 10% body surface area (BSA) involvement, Physician's Global Assessment (PGA) of at least moderate disease severity, and Psoriasis Area and Severity Index (PASI) ≥ 12 within three treatment periods. In period A, subjects received placebo or adalimumab at an initial dose of 80 mg at Week 0 followed by a dose of 40 mg every other week starting at Week 1. After 16 weeks of therapy, subjects who achieved at least a PASI 75 response at Week 16, defined as a PASI score improvement of at least 75% relative to baseline, entered period B and received open-label 40 mg adalimumab every other week. After 17 weeks of open-label therapy, subjects who maintained at least a PASI 75 response at Week 33 and were originally randomized to active therapy in period A were re-randomized in period C to receive 40 mg adalimumab every other week or placebo for an additional 19 weeks. Across all treatment groups the mean baseline PASI score was 19 and the baseline Physician's Global Assessment score ranged from "moderate" (53%) to "severe" (41%) to "very severe" (6%).
Study Ps-II evaluated 99 subjects randomized to adalimumab and 48 subjects randomized to placebo with chronic plaque psoriasis with ≥ 10% BSA involvement and PASI ≥ 12. Subjects received placebo, or an initial dose of 80 mg adalimumab at Week 0 followed by 40 mg every other week starting at Week 1 for 16 weeks. Across all treatment groups the mean baseline PASI score was 21 and the baseline PGA score ranged from "moderate" (41%) to "severe" (51%) to "very severe" (8%).
Studies Ps-I and II evaluated the proportion of subjects who achieved "clear" or "minimal" disease on the 6-point PGA scale and the proportion of subjects who achieved a reduction in PASI score of at least 75% (PASI 75) from baseline at Week 16 (see Table 16 and 17).
Additionally, Study Ps-I evaluated the proportion of subjects who maintained a PGA of "clear" or "minimal" disease or a PASI 75 response after Week 33 and on or before Week 52.
| Adalimumab 40 mg every other week | Placebo | |
|---|---|---|
| N = 814 | N = 398 | |
| PGA: Clear or minimal Clear = no plaque elevation, no scale, plus or minus hyperpigmentation or diffuse pink or red coloration
Minimal = possible but difficult to ascertain whether there is slight elevation of plaque above normal skin, plus or minus surface dryness with some white coloration, plus or minus up to red coloration |
506 (62%) | 17 (4%) |
| PASI 75 | 578 (71%) | 26 (7%) |
| Adalimumab 40 mg every other week | Placebo | |
|---|---|---|
| N = 99 | N = 48 | |
| PGA: Clear or minimal Clear = no plaque elevation, no scale, plus or minus hyperpigmentation or diffuse pink or red coloration
Minimal = possible but difficult to ascertain whether there is slight elevation of plaque above normal skin, plus or minus surface dryness with some white coloration, plus or minus up to red coloration |
70 (71%) | 5 (10%) |
| PASI 75 | 77 (78%) | 9 (19%) |
Additionally, in Study Ps-I, subjects on adalimumab who maintained a PASI 75 were re-randomized to adalimumab (N = 250) or placebo (N = 240) at Week 33. After 52 weeks of treatment with adalimumab, more subjects on adalimumab maintained efficacy when compared to subjects who were re-randomized to placebo based on maintenance of PGA of "clear" or "minimal" disease (68% vs. 28%) or a PASI 75 (79% vs. 43%).
A total of 347 stable responders participated in a withdrawal and retreatment evaluation in an open-label extension study. Median time to relapse (decline to PGA "moderate" or worse) was approximately 5 months. During the withdrawal period, no subject experienced transformation to either pustular or erythrodermic psoriasis. A total of 178 subjects who relapsed re-initiated treatment with 80 mg of adalimumab, then 40 mg every other week beginning at Week 1. At Week 16, 69% (123/178) of subjects had a response of PGA "clear" or "minimal".
A randomized, double-blind study (Study Ps-III) compared the efficacy and safety of adalimumab versus placebo in 217 adult subjects. Subjects in the study had to have chronic plaque psoriasis of at least moderate severity on the PGA scale, fingernail involvement of at least moderate severity on a 5-point Physician's Global Assessment of Fingernail Psoriasis (PGA-F) scale, a Modified Nail Psoriasis Severity Index (mNAPSI) score for the target-fingernail of ≥ 8, and either a BSA involvement of at least 10% or a BSA involvement of at least 5% with a total mNAPSI score for all fingernails of ≥ 20. Subjects received an initial dose of 80 mg adalimumab followed by 40 mg every other week (starting one week after the initial dose) or placebo for 26 weeks followed by open-label adalimumab treatment for an additional 26 weeks. This study evaluated the proportion of subjects who achieved "clear" or "minimal" assessment with at least a 2-grade improvement on the PGA-F scale and the proportion of subjects who achieved at least a 75% improvement from baseline in the mNAPSI score (mNAPSI 75) at Week 26.
At Week 26, a higher proportion of subjects in the adalimumab group than in the placebo group achieved the PGA-F endpoint. Furthermore, a higher proportion of subjects in the adalimumab group than in the placebo group achieved mNAPSI 75 at Week 26 (see Table 18).
| Endpoint | Adalimumab 40 mg every other week Subjects received 80 mg of adalimumab at Week 0, followed by 40 mg every other week starting at Week 1.
N = 109 |
Placebo N = 108 |
|---|---|---|
| PGA-F: ≥ 2-grade improvement and clear or minimal | 49% | 7% |
| mNAPSI 75 | 47% | 3% |
Nail pain was also evaluated and improvement in nail pain was observed in Study Ps-III.
Warning: Serious Infections and Malignancy
WARNING: SERIOUS INFECTIONS AND MALIGNANCY
See full prescribing information for complete boxed warning.
SERIOUS INFECTIONS (5.1, 6.1):
- Increased risk of serious infections leading to hospitalization or death, including tuberculosis (TB), bacterial sepsis, invasive fungal infections (such as histoplasmosis), and infections due to other opportunistic pathogens.
- Discontinue AMJEVITA if a patient develops a serious infection or sepsis during treatment.
- Perform test for latent TB; if positive, start treatment for TB prior to starting AMJEVITA.
- Monitor all patients for active TB during treatment, even if initial latent TB test is negative.
MALIGNANCY (5.2):
- Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers including adalimumab products.
- Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have occurred in adolescent and young adults with inflammatory bowel disease treated with TNF blockers including adalimumab products.
14.3 Clinical Studies in Psoriatic Arthritis
The safety and efficacy of adalimumab was assessed in two randomized, double-blind, placebo-controlled studies in 413 subjects with psoriatic arthritis (PsA). Upon completion of both studies, 383 subjects enrolled in an open-label extension study, in which 40 mg adalimumab was administered every other week.
Study PsA-I enrolled 313 adult subjects with moderately to severely active PsA (> 3 swollen and > 3 tender joints) who had an inadequate response to NSAID therapy in one of the following forms: (1) distal interphalangeal (DIP) involvement (N = 23); (2) polyarticular arthritis (absence of rheumatoid nodules and presence of plaque psoriasis) (N = 210); (3) arthritis mutilans (N = 1); (4) asymmetric PsA (N = 77); or (5) AS-like (N = 2). Subjects on MTX therapy (158 of 313 subjects) at enrollment (stable dose of ≤ 30 mg/week for > 1 month) could continue MTX at the same dose. Doses of adalimumab 40 mg or placebo every other week were administered during the 24-week double-blind period of the study.
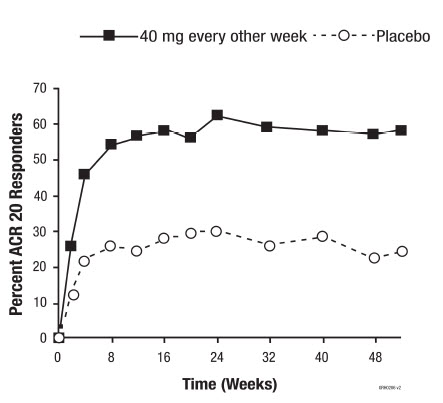
Compared to placebo, treatment with adalimumab resulted in improvements in the measures of disease activity (see Tables 8 and 9). Among subjects with PsA who received adalimumab, the clinical responses were apparent in some subjects at the time of the first visit (two weeks) and were maintained up to 88 weeks in the ongoing open-label study. Similar responses were seen in subjects with each of the subtypes of psoriatic arthritis, although few subjects were enrolled with the arthritis mutilans and ankylosing spondylitis-like subtypes. Responses were similar in subjects who were or were not receiving concomitant MTX therapy at baseline.
Subjects with psoriatic involvement of at least three percent body surface area (BSA) were evaluated for Psoriatic Area and Severity Index (PASI) responses. At 24 weeks, the proportions of subjects achieving a 75% or 90% improvement in the PASI were 59% and 42%, respectively, in the adalimumab group (N = 69), compared to 1% and 0%, respectively, in the placebo group (N = 69) (p < 0.001). PASI responses were apparent in some subjects at the time of the first visit (two weeks). Responses were similar in subjects who were or were not receiving concomitant MTX therapy at baseline.
| Placebo N = 162 |
Adalimumab p < 0.001 for all comparisons between adalimumab and placebo
N = 151 |
|
|---|---|---|
| ACR 20 | ||
| Week 12 | 14% | 58% |
| Week 24 | 15% | 57% |
| ACR 50 | ||
| Week 12 | 4% | 36% |
| Week 24 | 6% | 39% |
| ACR 70 | ||
| Week 12 | 1% | 20% |
| Week 24 | 1% | 23% |
| Placebo N = 162 |
Adalimumab p < 0.001 for adalimumab vs. placebo comparisons based on median changes
N = 151 |
|||
|---|---|---|---|---|
| Parameter: median | Baseline | 24 weeks | Baseline | 24 weeks |
| Number of tender joints Scale 0-78
|
23.0 | 17.0 | 20.0 | 5.0 |
| Number of swollen joints Scale 0-76
|
11.0 | 9.0 | 11.0 | 3.0 |
| Physician global assessment Visual analog scale; 0 = best, 100 = worst
|
53.0 | 49.0 | 55.0 | 16.0 |
| Patient global assessment | 49.5 | 49.0 | 48.0 | 20.0 |
| Pain | 49.0 | 49.0 | 54.0 | 20.0 |
| Disability index (HAQ) Disability Index of the Health Assessment Questionnaire; 0 = best, 3 = worst; measures the patient's ability to perform the following: dress/groom, arise, eat, walk, reach, grip, maintain hygiene, and maintain daily activity
|
1.0 | 0.9 | 1.0 | 0.4 |
| CRP (mg/dL) Normal range: 0-0.287 mg/dL
|
0.8 | 0.7 | 0.8 | 0.2 |
Similar results were seen in an additional, 12-week study in 100 subjects with moderate to severe psoriatic arthritis who had suboptimal response to DMARD therapy as manifested by ≥ 3 tender joints and ≥ 3 swollen joints at enrollment.
14.1 Clinical Studies in Rheumatoid Arthritis
The efficacy and safety of adalimumab were assessed in five randomized, double-blind studies in subjects ≥ 18 years of age with active rheumatoid arthritis (RA) diagnosed according to American College of Rheumatology (ACR) criteria. Subjects had at least 6 swollen and 9 tender joints. Adalimumab was administered subcutaneously in combination with methotrexate (MTX) (12.5 to 25 mg, Studies RA-I, RA-III and RA-V) or as monotherapy (Studies RA-II and RA-V) or with other disease-modifying anti-rheumatic drugs (DMARDs) (Study RA-IV).
Study RA-I evaluated 271 subjects who had failed therapy with at least one but no more than four DMARDs and had inadequate response to MTX. Doses of 20, 40 or 80 mg of adalimumab or placebo were given every other week for 24 weeks.
Study RA-II evaluated 544 subjects who had failed therapy with at least one DMARD. Doses of placebo, 20 or 40 mg of adalimumab were given as monotherapy every other week or weekly for 26 weeks.
Study RA-III evaluated 619 subjects who had an inadequate response to MTX. Subjects received placebo, 40 mg of adalimumab every other week with placebo injections on alternate weeks, or 20 mg of adalimumab weekly for up to 52 weeks. Study RA-III had an additional primary endpoint at 52 weeks of inhibition of disease progression (as detected by X-ray results). Upon completion of the first 52 weeks, 457 subjects enrolled in an open-label extension phase in which 40 mg of adalimumab was administered every other week for up to 5 years.
Study RA-IV assessed safety in 636 subjects who were either DMARD-naive or were permitted to remain on their pre-existing rheumatologic therapy provided that therapy was stable for a minimum of 28 days. Subjects were randomized to 40 mg of adalimumab or placebo every other week for 24 weeks.
Study RA-V evaluated 799 subjects with moderately to severely active RA of less than 3 years duration who were ≥ 18 years old and MTX naïve. Subjects were randomized to receive either MTX (optimized to 20 mg/week by Week 8), adalimumab 40 mg every other week or adalimumab/MTX combination therapy for 104 weeks. Subjects were evaluated for signs and symptoms, and for radiographic progression of joint damage. The median disease duration among subjects enrolled in the study was 5 months. The median MTX dose achieved was 20 mg.
14.11 Clinical Studies in Adults With Uveitis
The safety and efficacy of adalimumab were assessed in adult subjects with non-infectious intermediate, posterior and panuveitis excluding subjects with isolated anterior uveitis, in two randomized, double-masked, placebo-controlled studies (UV I and II). Subjects received placebo or adalimumab at an initial dose of 80 mg followed by 40 mg every other week starting one week after the initial dose. The primary efficacy endpoint in both studies was ´time to treatment failure´.
Treatment failure was a multi-component outcome defined as the development of new inflammatory chorioretinal and/or inflammatory retinal vascular lesions, an increase in anterior chamber (AC) cell grade or vitreous haze (VH) grade or a decrease in best corrected visual acuity (BCVA).
Study UV I evaluated 217 subjects with active uveitis while being treated with corticosteroids (oral prednisone at a dose of 10 to 60 mg/day). All subjects received a standardized dose of prednisone 60 mg/day at study entry followed by a mandatory taper schedule, with complete corticosteroid discontinuation by Week 15.
Study UV II evaluated 226 subjects with inactive uveitis while being treated with corticosteroids (oral prednisone 10 to 35 mg/day) at baseline to control their disease. Subjects subsequently underwent a mandatory taper schedule, with complete corticosteroid discontinuation by Week 19.
2.8 General Considerations for Administration
AMJEVITA is intended for use under the guidance and supervision of a physician. A patient may self-inject AMJEVITA or a caregiver may inject AMJEVITA using either the AMJEVITA prefilled SureClick autoinjector or prefilled syringe if a physician determines that it is appropriate, and with medical follow-up, as necessary, after proper training in subcutaneous injection technique.
AMJEVITA can be taken out of the refrigerator for 15 to 30 minutes before injecting to allow the liquid to come to room temperature. Do not remove the cap or cover while allowing it to reach room temperature. Carefully inspect the solution in the AMJEVITA prefilled SureClick autoinjector or prefilled syringe for particulate matter and discoloration prior to subcutaneous administration. If particulates and discolorations are noted, do not use the product. AMJEVITA does not contain preservatives; therefore, discard unused portions of drug remaining from the syringe.
Instruct patients using the AMJEVITA prefilled SureClick autoinjector or prefilled syringe to inject the full amount in the syringe, according to the directions provided in the Instructions for Use [see Instructions for Use].
Injections should occur at separate sites in the thigh or abdomen. Rotate injection sites and do not give injections into areas where the skin is tender, bruised, red or hard.
If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
Principal Display Panel 20 Mg Syringe Carton
20 mg/
0.4 mL
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-411-01
Not made with
natural rubber latex
For Use in Pediatric Patients
15 kg to < 30 kg
20 mg/0.4 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze.
Do not Shake. Protect from light.
(see side panel for additional storage information)
ATTENTION: Enclosed Medication Guide is required
for each patient.
Contains 1 Single-dose
Prefilled Syringe
CAUTION, Consult
Accompanying Documents
Do not re-use
Rx Only
AMGEN®
Principal Display Panel 40 Mg Syringe Carton
40
mg
/0.8 mL
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-410-01
Not made with
natural rubber latex
40 mg/0.8 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze.
Do not Shake. Protect from light.
(see side panel for additional storage information)
ATTENTION: Enclosed Medication Guide is required
for each patient.
Contains 1 Single-dose
Prefilled Syringe
CAUTION, Consult
Accompanying Documents
Do not re-use
Rx Only
AMGEN®
Principal Display Panel 80 Mg Syringe Carton
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-480-01
80 mg/
0.8 mL
80 mg/0.8 mL
For Subcutaneous Use Only.
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use. Do not Shake.
Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is
required for each patient.
Rx Only
Do not use
if carton seal is broken
Not made with
natural rubber latex
1
Single-dose
Prefilled Syringe
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
14.4 Clinical Studies in Ankylosing Spondylitis
The safety and efficacy of adalimumab 40 mg every other week was assessed in 315 adult subjects in a randomized, 24 week double-blind, placebo-controlled study in subjects with active ankylosing spondylitis (AS) who had an inadequate response to glucocorticoids, NSAIDs, analgesics, methotrexate or sulfasalazine. Active AS was defined as subjects who fulfilled at least two of the following three criteria: (1) a Bath AS disease activity index (BASDAI) score ≥ 4 cm, (2) a visual analog score (VAS) for total back pain ≥ 40 mm, and (3) morning stiffness ≥ 1 hour. The blinded period was followed by an open-label period during which subjects received adalimumab 40 mg every other week subcutaneously for up to an additional 28 weeks.
Improvement in measures of disease activity was first observed at Week 2 and maintained through 24 weeks as shown in Figure 2 and Table 11.
Responses of subjects with total spinal ankylosis (n = 11) were similar to those without total ankylosis.
At 12 weeks, the ASAS 20/50/70 responses were achieved by 58%, 38%, and 23%, respectively, of subjects receiving adalimumab, compared to 21%, 10%, and 5%, respectively, of subjects receiving placebo (p < 0.001). Similar responses were seen at Week 24 and were sustained in subjects receiving open-label adalimumab for up to 52 weeks.
A greater proportion of subjects treated with adalimumab (22%) achieved a low level of disease activity at 24 weeks (defined as a value < 20 [on a scale of 0 to 100 mm] in each of the four ASAS response parameters) compared to subjects treated with placebo (6%).
| Placebo N = 107 |
Adalimumab N = 208 |
|||
|---|---|---|---|---|
| Baseline mean | Week 24 mean | Baseline mean | Week 24 mean | |
| ASAS 20 Response Criteria Statistically significant for comparisons between adalimumab and placebo at Week 24
|
||||
| Patient's Global Assessment of Disease Activity Percent of subjects with at least a 20% and 10-unit improvement measured on a Visual Analog Scale (VAS) with 0 = "none" and 100 = "severe"
|
65 | 60 | 63 | 38 |
| Total back pain | 67 | 58 | 65 | 37 |
| Inflammation mean of questions 5 and 6 of BASDAI (defined in 'd')
|
6.7 | 5.6 | 6.7 | 3.6 |
| BASFI Bath Ankylosing Spondylitis Functional Index
|
56 | 51 | 52 | 34 |
| BASDAI Bath Ankylosing Spondylitis Disease Activity Index score
|
6.3 | 5.5 | 6.3 | 3.7 |
| BASMI Bath Ankylosing Spondylitis Metrology Index score
|
4.2 | 4.1 | 3.8 | 3.3 |
| Tragus to wall (cm) | 15.9 | 15.8 | 15.8 | 15.4 |
| Lumbar flexion (cm) | 4.1 | 4.0 | 4.2 | 4.4 |
| Cervical rotation (degrees) | 42.2 | 42.1 | 48.4 | 51.6 |
| Lumbar side flexion (cm) | 8.9 | 9.0 | 9.7 | 11.7 |
| Intermalleolar distance (cm) | 92.9 | 94.0 | 93.5 | 100.8 |
| CRP C-Reactive Protein (mg/dL)
|
2.2 | 2.0 | 1.8 | 0.6 |
A second randomized, multicenter, double-blind, placebo-controlled study of 82 subjects with ankylosing spondylitis showed similar results.
Subjects treated with adalimumab achieved improvement from baseline in the Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL) score (-3.6 vs. -1.1) and in the Short Form Health Survey (SF-36) Physical Component Summary (PCS) score (7.4 vs. 1.9) compared to placebo-treated subjects at Week 24.
14.10 Clinical Studies in Hidradenitis Suppurativa
Two randomized, double-blind, placebo-controlled studies (Studies HS-I and II) evaluated the safety and efficacy of adalimumab in a total of 633 adult subjects with moderate to severe hidradenitis suppurativa (HS) with Hurley Stage II or III disease and with at least 3 abscesses or inflammatory nodules. In both studies, subjects received placebo or adalimumab at an initial dose of 160 mg at Week 0, 80 mg at Week 2, and 40 mg every week starting at Week 4 and continued through Week 11. Subjects used topical antiseptic wash daily. Concomitant oral antibiotic use was allowed in Study HS-II.
Both studies evaluated Hidradenitis Suppurativa Clinical Response (HiSCR) at Week 12. HiSCR was defined as at least a 50% reduction in total abscess and inflammatory nodule count with no increase in abscess count and no increase in draining fistula count relative to baseline (see Table 19). Reduction in HS-related skin pain was assessed using a Numeric Rating Scale in subjects who entered the study with an initial baseline score of 3 or greater on a 11 point scale.
In both studies, a higher proportion of adalimumab- than placebo-treated subjects achieved HiSCR (see Table 19).
| HS Study I | HS Study II 19.3% of subjects in Study HS-II continued baseline oral antibiotic therapy during the study.
|
|||
|---|---|---|---|---|
| Placebo | Adalimumab 40 mg Weekly | Placebo | Adalimumab 40 mg Weekly | |
| Hidradenitis Suppurativa Clinical Response (HiSCR) | N = 154 40 (26%) |
N = 153 64 (42%) |
N=163 45 (28%) |
N=163 96 (59%) |
In both studies, from Week 12 to Week 35 (Period B), subjects who had received adalimumab were re-randomized to 1 of 3 treatment groups (adalimumab 40 mg every week, adalimumab 40 mg every other week, or placebo). Subjects who had been randomized to placebo were assigned to receive adalimumab 40 mg every week (Study HS-I) or placebo (Study HS-II).
During Period B, flare of HS, defined as ≥ 25% increase from baseline in abscesses and inflammatory nodule counts and with a minimum of 2 additional lesions, was documented in 22 (22%) of the 100 subjects who were withdrawn from adalimumab treatment following the primary efficacy timepoint in two studies.
Principal Display Panel 40 Mg Autoinjector Carton
40 mg/
0.8 mL
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-400-01
SureClick®
Prefilled Autoinjector
Not made with
natural rubber latex
40 mg/0.8 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze.
Do not Shake. Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is
required for each patient.
Contains 1 Single-dose
Prefilled Autoinjector
CAUTION, Consult
Accompanying Documents
Do not re-use
Rx Only
AMGEN®
Principal Display Panel 80 Mg Autoinjector Carton
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-481-01
80 mg/
0.8 mL
80 mg/0.8 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use.
Do not Shake. Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is
required for each patient.
Rx Only
Not made with
natural rubber latex
1
Single-dose Prefilled
SureClick® Autoinjector
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
14.5 Clinical Studies in Adults With Crohn's Disease
The safety and efficacy of multiple doses of adalimumab were assessed in adult subjects with moderately to severely active Crohn's disease, CD, (Crohn's Disease Activity Index (CDAI) ≥ 220 and ≤ 450) in randomized, double-blind, placebo-controlled studies. Concomitant stable doses of aminosalicylates, corticosteroids, and/or immunomodulatory agents were permitted, and 79% of subjects continued to receive at least one of these medications.
Induction of clinical remission (defined as CDAI < 150) was evaluated in two studies. In Study CD-I, 299 TNF-blocker naïve subjects were randomized to one of four treatment groups: the placebo group received placebo at Weeks 0 and 2, the 160/80 group received 160 mg adalimumab at Week 0 and 80 mg at Week 2, the 80/40 group received 80 mg at Week 0 and 40 mg at Week 2, and the 40/20 group received 40 mg at Week 0 and 20 mg at Week 2. Clinical results were assessed at Week 4.
In the second induction study, Study CD-II, 325 subjects who had lost response to, or were intolerant to, previous infliximab therapy were randomized to receive either 160 mg adalimumab at Week 0 and 80 mg at Week 2, or placebo at Weeks 0 and 2. Clinical results were assessed at Week 4.
Maintenance of clinical remission was evaluated in Study CD-III. In this study, 854 subjects with active disease received open-label adalimumab, 80 mg at Week 0 and 40 mg at Week 2. Subjects were then randomized at Week 4 to 40 mg adalimumab every other week, 40 mg adalimumab every week, or placebo. The total study duration was 56 weeks. Subjects in clinical response (decrease in CDAI ≥ 70) at Week 4 were stratified and analyzed separately from those not in clinical response at Week 4.
Principal Display Panel 10 Mg/0.2 Ml Syringe Carton
AMGEN®
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-413-01
10 mg/
0.2 mL
1
Single-dose
Prefilled Syringe
10 mg/0.2 mL
For Subcutaneous Use Only.
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use. Do not Shake.
Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is required
for each patient.
Rx Only
Do not use
if carton seal is broken
Not made with
natural rubber latex
For Use in Pediatric Patients
10 kg to < 15 kg
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
Principal Display Panel 20 Mg/0.2 Ml Syringe Carton
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-399-01
20 mg/
0.2 mL
20 mg/0.2 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use. Do not Shake.
Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is required
for each patient.
Rx Only
Do not use
if carton seal is broken
Not made with
natural rubber latex
1
Single-dose
Prefilled Syringe
For Use in Pediatric Patients
15 kg to < 30 kg
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
Principal Display Panel 40 Mg/0.4 Ml Syringe Carton
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-479-01
40 mg/
0.4 mL
40 mg/0.4 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use. Do not Shake.
Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is required
for each patient.
Rx Only
Do not use
if carton seal is broken
Not made with
natural rubber latex
1
Single-dose
Prefilled Syringe
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
14.2 Clinical Studies in Juvenile Idiopathic Arthritis
The safety and efficacy of adalimumab was assessed in two studies (Studies JIA-I and JIA-II) in subjects with active polyarticular juvenile idiopathic arthritis (JIA).
14.7 Clinical Studies in Adults With Ulcerative Colitis
The safety and efficacy of adalimumab were assessed in adult subjects with moderately to severely active ulcerative colitis (Mayo score 6 to 12 on a 12 point scale, with an endoscopy subscore of 2 to 3 on a scale of 0 to 3) despite concurrent or prior treatment with immunosuppressants such as corticosteroids, azathioprine, or 6-MP in two randomized, double-blind, placebo-controlled clinical studies (Studies UC-I and UC-II). Both studies enrolled TNF-blocker naïve subjects, but Study UC-II also allowed entry of subjects who lost response to or were intolerant to TNF-blockers. Forty percent (40%) of subjects enrolled in Study UC-II had previously used another TNF-blocker.
Concomitant stable doses of aminosalicylates and immunosuppressants were permitted. In Studies UC-I and II, subjects were receiving aminosalicylates (69%), corticosteroids (59%) and/or azathioprine or 6-MP (37%) at baseline. In both studies, 92% of subjects received at least one of these medications.
Induction of clinical remission (defined as Mayo score ≤ 2 with no individual subscores > 1) at Week 8 was evaluated in both studies. Clinical remission at Week 52 and sustained clinical remission (defined as clinical remission at both Weeks 8 and 52) were evaluated in Study UC-II.
In Study UC-I, 390 TNF-blocker naïve subjects were randomized to one of three treatment groups for the primary efficacy analysis. The placebo group received placebo at Weeks 0, 2, 4 and 6. The 160/80 group received 160 mg adalimumab at Week 0 and 80 mg at Week 2, and the 80/40 group received 80 mg adalimumab at Week 0 and 40 mg at Week 2. After Week 2, subjects in both adalimumab treatment groups received 40 mg every other week.
In Study UC-II, 518 subjects were randomized to receive either adalimumab 160 mg at Week 0, 80 mg at Week 2, and 40 mg every other week starting at Week 4 through Week 50, or placebo starting at Week 0 and every other week through Week 50. Corticosteroid taper was permitted starting at Week 8.
In both Studies UC-I and UC-II, a greater percentage of the subjects treated with 160/80 mg of adalimumab compared to subjects treated with placebo achieved induction of clinical remission. In Study UC-II, a greater percentage of the subjects treated with 160/80 mg of adalimumab compared to subjects treated with placebo achieved sustained clinical remission (clinical remission at both Weeks 8 and 52) (see Table 15).
| Study UC-I | Study UC-II | |||||
|---|---|---|---|---|---|---|
| Placebo N = 130 |
Adalimumab 160/80 mg N = 130 |
Treatment Difference (95% CI) |
Placebo N = 246 |
Adalimumab 160/80 mg N = 248 |
Treatment Difference (95% CI) |
|
| Clinical remission is defined as Mayo score ≤ 2 with no individual subscores > 1. CI = Confidence interval |
||||||
| Induction of Clinical Remission (Clinical Remission at Week 8) | 9.2% | 18.5% | 9.3% p < 0.05 for adalimumab vs. placebo pairwise comparison of proportions
(0.9%, 17.6%) |
9.3% | 16.5% | 7.2%
(1.2%, 12.9%) |
| Sustained Clinical Remission (Clinical Remission at both Weeks 8 and 52) | N/A | N/A | N/A | 4.1% | 8.5% | 4.4%
(0.1%, 8.6%) |
In Study UC-I, there was no statistically significant difference in clinical remission observed between the adalimumab 80/40 mg group and the placebo group at Week 8.
In Study UC-II, 17.3% (43/248) in the adalimumab group were in clinical remission at Week 52 compared to 8.5% (21/246) in the placebo group (treatment difference: 8.8%; 95% confidence interval (CI): [2.8%, 14.5%]; p < 0.05).
In the subgroup of subjects in Study UC-II with prior TNF-blocker use, the treatment difference for induction of clinical remission appeared to be lower than that seen in the whole study population, and the treatment differences for sustained clinical remission and clinical remission at Week 52 appeared to be similar to those seen in the whole study population. The subgroup of subjects with prior TNF-blocker use achieved induction of clinical remission at 9% (9/98) in the adalimumab group versus 7% (7/101) in the placebo group, and sustained clinical remission at 5% (5/98) in the adalimumab group versus 1% (1/101) in the placebo group. In the subgroup of subjects with prior TNF-blocker use, 10% (10/98) were in clinical remission at Week 52 in the adalimumab group versus 3% (3/101) in the placebo group.
5.7 Increased Risk of Infection When Used With Anakinra
Concurrent use of anakinra (an interleukin-1 antagonist) and another TNF-blocker, was associated with a greater proportion of serious infections and neutropenia and no added benefit compared with the TNF-blocker alone in patients with RA. Therefore, the combination of AMJEVITA and anakinra is not recommended [see Drug Interactions (7.2)].
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies of adalimumab products have not been conducted to evaluate the carcinogenic potential or its effect on fertility.
14.12 Clinical Studies in Pediatric Subjects With Uveitis
The safety and efficacy of adalimumab were assessed in a randomized, double-masked, placebo-controlled study of 90 pediatric subjects from 2 to < 18 years of age with active JIA-associated non-infectious uveitis (PUV-I). Subjects received either placebo or 20 mg adalimumab (if < 30 kg) or 40 mg adalimumab (if ≥ 30 kg) every other week in combination with a dose of methotrexate. Concomitant dosages of corticosteroids were permitted at study entry followed by a mandatory reduction in topical corticosteroids within 3 months.
The primary endpoint was 'time to treatment failure'. The criteria determining treatment failure were worsening or sustained non-improvement in ocular inflammation, or worsening of ocular co-morbidities.
5.11 Increased Risk of Infection When Used With Abatacept
In controlled trials, the concurrent administration of TNF-blockers and abatacept was associated with a greater proportion of serious infections than the use of a TNF-blocker alone; the combination therapy, compared to the use of a TNF-blocker alone, has not demonstrated improved clinical benefit in the treatment of RA. Therefore, the combination of abatacept with TNF-blockers including AMJEVITA is not recommended [see Drug Interactions (7.2)].
Principal Display Panel 40 Mg/0.4 Ml Autoinjector Carton
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-482-01
40 mg/
0.4 mL
40 mg/0.4 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use.
Do not Shake. Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide
is required for each patient.
Rx Only
Not made with
natural rubber latex
1
Single-dose Prefilled
SureClick® Autoinjector
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
14.6 Clinical Studies in Pediatric Subjects With Crohn's Disease
A randomized, double-blind, 52-week clinical study of 2 dose concentrations of adalimumab (Study PCD-I) was conducted in 192 pediatric subjects (6 to 17 years of age) with moderately to severely active Crohn's disease (defined as Pediatric Crohn's Disease Activity Index (PCDAI) score > 30). Enrolled subjects had over the previous two year period an inadequate response to corticosteroids or an immunomodulator (i.e., azathioprine, 6-mercaptopurine, or methotrexate). Subjects who had previously received a TNF blocker were allowed to enroll if they had previously had loss of response or intolerance to that TNF blocker.
Subjects received open-label induction therapy at a dose based on their body weight (≥ 40 kg and < 40 kg). Subjects weighing ≥ 40 kg received 160 mg (at Week 0) and 80 mg (at Week 2). Subjects weighing < 40 kg received 80 mg (at Week 0) and 40 mg (at Week 2). At Week 4, subjects within each body weight category (≥ 40 kg and < 40 kg) were randomized 1:1 to one of two maintenance dose regimens (high dose and low dose). The high dose was 40 mg every other week for subjects weighing ≥ 40 kg and 20 mg every other week for subjects weighing < 40 kg. The low dose was 20 mg every other week for subjects weighing ≥ 40 kg and 10 mg every other week for subjects weighing < 40 kg.
Concomitant stable dosages of corticosteroids (prednisone dosage ≤ 40 mg/day or equivalent) and immunomodulators (azathioprine, 6-mercaptopurine, or methotrexate) were permitted throughout the study.
At Week 12, subjects who experienced a disease flare (increase in PCDAI of ≥ 15 from Week 4 and absolute PCDAI > 30) or who were non-responders (did not achieve a decrease in the PCDAI of ≥ 15 from baseline for 2 consecutive visits at least 2 weeks apart) were allowed to dose-escalate (i.e., switch from blinded every other week dosing to blinded every week dosing); subjects who dose-escalated were considered treatment failures.
At baseline, 38% of subjects were receiving corticosteroids, and 62% of subjects were receiving an immunomodulator. Forty-four percent (44%) of subjects had previously lost response or were intolerant to a TNF blocker. The median baseline PCDAI score was 40.
Of the 192 subjects total, 188 subjects completed the 4 week induction period, 152 subjects completed 26 weeks of treatment, and 124 subjects completed 52 weeks of treatment. Fifty-one percent (51%) (48/95) of subjects in the low maintenance dose group dose-escalated, and 38% (35/93) of subjects in the high maintenance dose group dose-escalated.
At Week 4, 28% (52/188) of subjects were in clinical remission (defined as PCDAI ≤ 10).
The proportions of subjects in clinical remission (defined as PCDAI ≤ 10) and clinical response (defined as reduction in PCDAI of at least 15 points from baseline) were assessed at Weeks 26 and 52.
At both Weeks 26 and 52, the proportion of subjects in clinical remission and clinical response was numerically higher in the high dose group compared to the low dose group (Table 14). The recommended maintenance regimen is 20 mg every other week for subjects weighing < 40 kg and 40 mg every other week for subjects weighing ≥ 40 kg. Every week dosing is not the recommended maintenance dosing regimen [see Dosage and Administration (2.3)].
| Low Maintenance Dose The low maintenance dose was 20 mg every other week for subjects weighing ≥ 40 kg and 10 mg every other week for subjects weighing < 40 kg.
(20 or 10 mg every other week) N = 95 |
High Maintenance Dose The high maintenance dose was 40 mg every other week for subjects weighing ≥ 40 kg and 20 mg every other week for subjects weighing < 40 kg.
(40 or 20 mg every other week) N = 93 |
|
|---|---|---|
| Week 26 | ||
| Clinical remission Clinical remission defined as PCDAI ≤ 10.
|
28% | 39% |
| Clinical response Clinical response defined as reduction in PCDAI of at least 15 points from baseline.
|
48% | 59% |
| Week 52 | ||
| Clinical remission | 23% | 33% |
| Clinical response | 28% | 42% |
2.6 Recommended Dosage in Plaque Psoriasis Or Adults With Uveitis
The recommended subcutaneous dosage of AMJEVITA for adult patients with plaque psoriasis (Ps) or uveitis (UV) [see Indications and Usage (1.7, 1.9)] is an initial dose of 80 mg, followed by 40 mg given every other week starting one week after the initial dose. The use of adalimumab products in moderate to severe chronic Ps beyond one year has not been evaluated in controlled clinical studies.
2.3 Recommended Dosage in Juvenile Idiopathic Arthritis Or Pediatric Patients With Uveitis
The recommended subcutaneous dosage of AMJEVITA for pediatric patients 2 years of age and older with polyarticular juvenile idiopathic arthritis (JIA) or pediatric uveitis [see Indications and Usage (1.2, 1.9)], based on weight, is shown below. MTX, glucocorticoids, NSAIDs, and/or analgesics may be continued during treatment with AMJEVITA.
| Pediatric Weight (2 Years of Age and Older) | Recommended Dosage |
|---|---|
| 10 kg (22 lbs) to less than 15 kg (33 lbs) | 10 mg every other week |
| 15 kg (33 lbs) to less than 30 kg (66 lbs) | 20 mg every other week |
| 30 kg (66 lbs) and greater | 40 mg every other week |
Adalimumab products have not been studied in patients with polyarticular JIA or pediatric uveitis less than 2 years of age or in patients with a weight below 10 kg.
2.2 Recommended Dosage in Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
The recommended subcutaneous dosage of AMJEVITA for adult patients with rheumatoid arthritis (RA), psoriatic arthritis (PsA), or ankylosing spondylitis (AS) [see Indication and Usage (1.1, 1.3, 1.4)] is 40 mg administered every other week. Methotrexate (MTX), other non-biologic DMARDs, glucocorticoids, nonsteroidal anti-inflammatory drugs (NSAIDs), and/or analgesics may be continued during treatment with AMJEVITA. In the treatment of RA, some patients not taking concomitant MTX may derive additional benefit from increasing the dosage of AMJEVITA to 40 mg every week or 80 mg every other week.
Structured Label Content
Section 42229-5 (42229-5)
SERIOUS INFECTIONS
Patients treated with adalimumab products, including AMJEVITA, are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
Discontinue AMJEVITA if a patient develops a serious infection or sepsis.
Reported infections include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients with TB have frequently presented with disseminated or extrapulmonary disease. Test patients for latent TB before AMJEVITA use and during therapy. Initiate treatment for latent TB prior to AMJEVITA use.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. Consider empiric anti-fungal therapy in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral and other infections due to opportunistic pathogens, including Legionella and Listeria.
Carefully consider the risks and benefits of treatment with AMJEVITA prior to initiating therapy in patients with chronic or recurrent infection.
Monitor patients closely for the development of signs and symptoms of infection during and after treatment with AMJEVITA, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Section 42231-1 (42231-1)
| MEDICATION GUIDE AMJEVITA® (am-jeh-vee'-tah) (adalimumab-atto) injection, for subcutaneous use |
||
|---|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 10/2025 | |
| Read the Medication Guide that comes with AMJEVITA before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment. | ||
|
What is the most important information I should know about AMJEVITA?
Before starting AMJEVITA, tell your doctor if you:
|
||
|
|
|
AMJEVITA can make you more likely to get infections or make any infection that you may have worse. Cancer
|
||
|
What is AMJEVITA?
AMJEVITA is a medicine called a Tumor Necrosis Factor (TNF) blocker. AMJEVITA is used:
|
||
|
What should I tell my doctor before taking AMJEVITA?
AMJEVITA may not be right for you. Before starting AMJEVITA, tell your doctor about all of your medical conditions, including if you:
Especially tell your doctor if you use:
|
||
How should I take AMJEVITA?
|
||
|
What are the possible side effects of AMJEVITA?
AMJEVITA can cause serious side effects, including: See "What is the most important information I should know about AMJEVITA?"
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
|
||
|
|
|
The most common side effects of AMJEVITA include:
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store AMJEVITA?
|
||
|
General information about the safe and effective use of AMJEVITA
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use AMJEVITA for a condition for which it was not prescribed. Do not give AMJEVITA to other people, even if they have the same condition. It may harm them. This Medication Guide summarizes the most important information about AMJEVITA. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about AMJEVITA that is written for health professionals. |
||
|
What are the ingredients in AMJEVITA?
Active ingredient: adalimumab-atto Inactive ingredients: glacial acetic acid (or L-lactic acid), polysorbate 80, sucrose and Water for Injection. Sodium hydroxide is added as necessary to adjust the pH to 5.2. AMJEVITA® (adalimumab-atto) Manufactured by: Amgen Inc., One Amgen Center Drive, Thousand Oaks, CA 91320-1799 U.S.A U.S. License Number 1080 © 2022–2025 Amgen Inc. All rights reserved.1xxxxxx – v10 For more information call 1-800-77-AMGEN (1-800-772-6436). |
Section 43683-2 (43683-2)
Section 44425-7 (44425-7)
Storage and Stability
Do not use beyond the expiration date on the container. AMJEVITA must be refrigerated at 36°F to 46°F (2°C to 8°C). DO NOT FREEZE. Do not use if frozen even if it has been thawed.
Store in original carton until time of administration to protect from light.
If needed, for example when traveling, AMJEVITA may be stored at room temperature up to a maximum of 77°F (25°C) for a period of up to 14 days, with protection from light. AMJEVITA should be discarded if not used within the 14-day period. Record the date when AMJEVITA is first removed from the refrigerator in the spaces provided on the carton.
Do not store AMJEVITA in extreme heat or cold.
1.9 Uveitis
AMJEVITA is indicated for the treatment of non-infectious intermediate, posterior, and panuveitis in adults and pediatric patients 2 years of age and older.
15 References (15 REFERENCES)
- National Cancer Institute. Surveillance, Epidemiology, and End Results Database (SEER) Program. SEER Incidence Crude Rates, 17 Registries, 2000-2007.
10. Overdosage (10. OVERDOSAGE)
Doses up to 10 mg/kg have been administered to patients in clinical trials without evidence of dose-limiting toxicities. In case of overdosage, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions or effects and appropriate symptomatic treatment instituted immediately.
Consider contacting the Poison Help line (1-800-222-1222) or medical toxicologist for additional overdose management recommendations.
11 Description (11 DESCRIPTION)
Adalimumab-atto is a tumor necrosis factor blocker. Adalimumab-atto is a recombinant human IgG1 monoclonal antibody with human derived heavy and light chain variable regions and human IgG1:k constant regions. Adalimumab-atto is produced by recombinant DNA technology in a mammalian cell (Chinese Hamster Ovary (CHO)) expression system and is purified by a process that includes specific viral inactivation and removal steps. It consists of 1330 amino acids and has a molecular weight of approximately 148 kilodaltons.
AMJEVITA® (adalimumab-atto) injection is supplied as a sterile, preservative-free solution for subcutaneous administration. The drug product is supplied as either a single-dose, prefilled SureClick autoinjector, or as a single-dose, 1 mL prefilled glass syringe. Enclosed within the autoinjector is a single-dose, 1 mL prefilled glass syringe. The solution of AMJEVITA is clear, colorless to slightly yellow, with a pH of about 5.2.
Each 80 mg/0.8 mL prefilled syringe or prefilled autoinjector delivers 0.8 mL (80 mg) of drug product. Each 0.8 mL of AMJEVITA is formulated with L-lactic acid (1.7 mg), polysorbate 80 (0.8 mg), sodium hydroxide for pH adjustment, sucrose (67 mg), and Water for Injection, USP, pH 5.2.
Each 40 mg/0.8 mL prefilled syringe or prefilled autoinjector delivers 0.8 mL (40 mg) of drug product. Each 0.8 mL of AMJEVITA is formulated with glacial acetic acid (0.48 mg), polysorbate 80 (0.8 mg), sodium hydroxide for pH adjustment, sucrose (72 mg), and Water for Injection, USP, pH 5.2.
Each 40 mg/0.4 mL prefilled syringe or prefilled autoinjector delivers 0.4 mL (40 mg) of drug product. Each 0.4 mL of AMJEVITA is formulated with L-lactic acid (0.9 mg), polysorbate 80 (0.4 mg), sodium hydroxide for pH adjustment, sucrose (34 mg), and Water for Injection, USP, pH 5.2.
Each 20 mg/0.4 mL prefilled syringe delivers 0.4 mL (20 mg) of drug product. Each 0.4 mL of AMJEVITA is formulated with glacial acetic acid (0.24 mg), polysorbate 80 (0.4 mg), sodium hydroxide for pH adjustment, sucrose (36 mg), and Water for Injection, USP, pH 5.2.
Each 20 mg/0.2 mL prefilled syringe delivers 0.2 mL (20 mg) of drug product. Each 0.2 mL of AMJEVITA is formulated with L-lactic acid (0.4 mg), polysorbate 80 (0.2 mg), sodium hydroxide for pH adjustment, sucrose (17 mg), and Water for Injection, USP, pH 5.2.
Each 10 mg/0.2 mL prefilled syringe delivers 0.2 mL (10 mg) of drug product. Each 0.2 mL of AMJEVITA is formulated with glacial acetic acid (0.12 mg), polysorbate 80 (0.2 mg), sodium hydroxide for pH adjustment, sucrose (18 mg), and Water for Injection, USP, pH 5.2.
5.2 Malignancies
Consider the risks and benefits of TNF-blocker treatment including AMJEVITA prior to initiating therapy in patients with a known malignancy other than a successfully treated non-melanoma skin cancer (NMSC) or when considering continuing a TNF blocker in patients who develop a malignancy.
5.9 Autoimmunity
Treatment with adalimumab products may result in the formation of autoantibodies and, rarely, in the development of a lupus-like syndrome or autoimmune hepatitis [see Adverse Reactions (6.1, 6.3)]. If a patient develops symptoms and findings suggestive of a lupus-like syndrome or autoimmune hepatitis following treatment with AMJEVITA, discontinue treatment and evaluate the patient.
7.1 Methotrexate
Adalimumab has been studied in rheumatoid arthritis (RA) patients taking concomitant methotrexate (MTX). Although MTX reduced the apparent clearance of adalimumab, the data do not suggest the need for dose adjustment of either AMJEVITA or MTX [see Clinical Pharmacology (12.3)].
5.8 Heart Failure
Cases of worsening congestive heart failure (CHF) and new onset CHF have been reported with TNF blockers. Cases of worsening CHF have also been observed with adalimumab products. Adalimumab products have not been formally studied in patients with CHF; however, in clinical trials of another TNF blocker, a higher rate of serious CHF-related adverse reactions was observed. Exercise caution when using AMJEVITA in patients who have heart failure and monitor them carefully.
7.3 Live Vaccines
Avoid the use of live vaccines with AMJEVITA [see Warnings and Precautions (5.10)].
8.4 Pediatric Use
The safety and effectiveness of AMJEVITA have not been established in pediatric patients with psoriatic arthritis, ankylosing spondylitis, or plaque psoriasis.
The safety and effectiveness of AMJEVITA have been established for:
- reducing signs and symptoms of moderately to severely active polyarticular JIA in pediatric patients 2 years of age and older.
- the treatment of moderately to severely active Crohn's disease in pediatric patients 6 years of age and older.
- the treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older.
- the treatment of non-infectious intermediate, posterior, and panuveitis in pediatric patients 2 years of age and older.
A pediatric assessment for AMJEVITA demonstrates that AMJEVITA is safe and effective for pediatric patients in an indication for which HUMIRA (adalimumab) is approved. However, AMJEVITA is not approved for such indication due to marketing exclusivity for HUMIRA (adalimumab).
Due to their inhibition of TNFα, adalimumab products administered during pregnancy could affect immune response in the in utero-exposed newborn and infant. Data from eight infants exposed to adalimumab in utero suggest adalimumab crosses the placenta [see Use in Specific Populations (8.1)]. The clinical significance of elevated adalimumab concentrations in infants is unknown. The safety of administering live or live-attenuated vaccines in exposed infants is unknown. Risks and benefits should be considered prior to vaccinating (live or live-attenuated) exposed infants.
Post-marketing cases of lymphoma, including hepatosplenic T-cell lymphoma and other malignancies, some fatal, have been reported among children, adolescents, and young adults who received treatment with TNF-blockers including adalimumab products [see Warnings and Precautions (5.2)].
8.5 Geriatric Use
In clinical studies of RA (Studies RA-I, RA-II, RA-III, and RA-IV), a total of 519 subjects 65 years of age and older, including 107 subjects 75 years of age and older, received adalimumab. No overall difference in effectiveness was observed between these subjects and younger adult subjects.
The frequency of serious infection and malignancy among adalimumab-treated subjects 65 years of age and older was higher than for those less than 65 years of age. Consider the benefits and risks of AMJEVITA in patients 65 years of age and older. In patients treated with AMJEVITA, closely monitor for the development of infection or malignancy [see Warnings and Precautions (5.1, 5.2)].
5.10 Immunizations
In a placebo-controlled clinical trial of patients with RA, no difference was detected in anti-pneumococcal antibody response between adalimumab and placebo treatment groups when the pneumococcal polysaccharide vaccine and influenza vaccine were administered concurrently with adalimumab. Similar proportions of subjects developed protective levels of anti-influenza antibodies between adalimumab and placebo treatment groups; however, titers in aggregate to influenza antigens were moderately lower in patients receiving adalimumab. The clinical significance of this is unknown. Patients on AMJEVITA may receive concurrent vaccinations, except for live vaccines. No data are available on the secondary transmission of infection by live vaccines in patients receiving adalimumab products.
It is recommended that pediatric patients, if possible, be brought up to date with all immunizations in agreement with current immunization guidelines prior to initiating AMJEVITA therapy. Patients on AMJEVITA may receive concurrent vaccinations, except for live vaccines.
The safety of administering live or live-attenuated vaccines in infants exposed to adalimumab products in utero is unknown. Risks and benefits should be considered prior to vaccinating (live or live-attenuated) exposed infants [see Use in Specific Populations (8.1 , 8.4)].
6.2 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of adalimumab or of other adalimumab products.
There are two assays that have been used to measure anti-adalimumab antibodies. With the ELISA, antibodies to adalimumab could be detected only when serum adalimumab concentrations were < 2 mcg/mL. The ECL assay can detect anti-adalimumab antibody titers independent of adalimumab concentrations in the serum samples. The incidence of anti-adalimumab antibody (AAA) development in patients treated with adalimumab are presented in Table 2.
| Indications | Study Duration | Anti-Adalimumab Antibody Incidence by ELISA (n/N) | Anti-Adalimumab Antibody Incidence by ECL Assay (n/N) | ||
|---|---|---|---|---|---|
| In all patients who received adalimumab | In patients with serum adalimumab concentrations < 2 mcg/mL | ||||
| n: number of patients with anti-adalimumab antibody; NR: not reported; NA: Not applicable (not performed) | |||||
| Rheumatoid Arthritis In patients receiving concomitant methotrexate (MTX), the incidence of anti-adalimumab antibody was 1% compared to 12% with adalimumab monotherapy
|
6 to 12 months | 5% (58/1062) | NR | NA | |
| Juvenile Idiopathic Arthritis (JIA) | 4 to 17 years of age In patients receiving concomitant MTX, the incidence of anti-adalimumab antibody was 6% compared to 26% with adalimumab monotherapy
|
48 weeks | 16% (27/171) | NR | NA |
| 2 to 4 years of age or ≥ 4 years of age and weighing < 15 kg | 24 weeks | 7% (1/15) This patient received concomitant MTX
|
NR | NA | |
| Psoriatic Arthritis In patients receiving concomitant MTX, the incidence of antibody development was 7% compared to 1% in RA
|
48 weeks Subjects enrolled after completing 2 previous studies of 24 weeks or 12 weeks of treatments
|
13% (24/178) | NR | NA | |
| Ankylosing Spondylitis | 24 weeks | 9% (16/185) | NR | NA | |
| Adult Crohn's Disease | 56 weeks | 3% (7/269) | 8% (7/86) | NA | |
| Pediatric Crohn's Disease | 52 weeks | 3% (6/182) | 10% (6/58) | NA | |
| Adult Ulcerative Colitis | 52 weeks | 5% (19/360) | 21% (19/92) | NA | |
| Plaque Psoriasis In plaque psoriasis patients who were on adalimumab monotherapy and subsequently withdrawn from the treatment, the rate of antibodies to adalimumab after retreatment was similar to the rate observed prior to withdrawal
|
Up to 52 weeks One 12-week Phase 2 study and one 52-week Phase 3 study
|
8% (77/920) | 21% (77/372) | NA | |
| Hidradenitis Suppurativa | 36 weeks | 7% (30/461) | 28% (58/207) Among subjects in the two Phase 3 studies who stopped adalimumab treatment for up to 24 weeks and in whom adalimumab serum levels subsequently declined to < 2 mcg/mL (approximately 22% of total subjects studied)
|
61% (272/445 No apparent association between antibody development and safety was observed
|
|
| Non-infectious Uveitis | 52 weeks | 5% (12/249) | 21% (12/57) | 40% (99/249) No correlation of antibody development to safety or efficacy outcomes was observed
|
1.5 Crohn's Disease
AMJEVITA is indicated for the treatment of moderately to severely active Crohn's disease in adults and pediatric patients 6 years of age and older.
4 Contraindications (4 CONTRAINDICATIONS)
None.
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Malignancies [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Hepatitis B Virus Reactivation [see Warnings and Precautions (5.4)]
- Neurologic Reactions [see Warnings and Precautions (5.5)]
- Hematological Reactions [see Warnings and Precautions (5.6)]
- Heart Failure [see Warnings and Precautions (5.8)]
- Autoimmunity [see Warnings and Precautions (5.9)]
7 Drug Interactions (7 DRUG INTERACTIONS)
1.7 Plaque Psoriasis
AMJEVITA is indicated for the treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy, and when other systemic therapies are medically less appropriate. AMJEVITA should only be administered to patients who will be closely monitored and have regular follow-up visits with a physician [see Warnings and Precautions (5)].
Instructions for Use (INSTRUCTIONS FOR USE)
AMJEVITA® (am-jeh-vee'-tah)
(adalimumab-atto)
injection, for subcutaneous use
40 mg/0.8 mL
single-dose prefilled SureClick® autoinjector
This Instructions for Use contains information on how to inject AMJEVITA with a SureClick autoinjector.
If your healthcare provider decides that you or a caregiver may be able to give your injections of AMJEVITA at home, you should receive training on the right way to prepare and inject AMJEVITA. Do not try to inject yourself until you have been shown the right way to give the injections by your healthcare provider or nurse.
The medicine in the AMJEVITA autoinjector is for injection under the skin (subcutaneous injection). See the AMJEVITA Medication Guide for information about AMJEVITA.
| Getting to know your prefilled autoinjector |
1. Important Information You Need to Know Before Injecting AMJEVITA
- It is important that you do not try to give the injection until you have fully read and understood this Instructions for Use.
- Do not use the autoinjector if the carton is damaged or the seal is broken.
- Do not use the autoinjector after the expiration date on the label.
- Do not shake the autoinjector.
- Do not remove the yellow cap from the autoinjector until you are ready to inject.
- Do not use the autoinjector if it has been frozen.
- Do not use the autoinjector if it has been dropped on a hard surface. Part of the autoinjector may be broken even if you cannot see the break. Use a new autoinjector, and call 1-800-77-AMGEN (1-800-772-6436).
- The autoinjector is not made with natural rubber latex.
| Important: Keep the autoinjector and sharps disposal container out of the sight and reach of children. |
Frequently asked questions:
For additional information and answers to frequently asked questions, visit www.amjevita.com.
Where to get help:
If you want more information or help using AMJEVITA:
- Contact your healthcare provider,
- Visit www.amjevita.com, or
- Call 1-800-77-AMGEN (1-800-772-6436)
2. Storing and Preparing to Inject AMJEVITA
2a Refrigerate the autoinjector carton until you are ready to use it.
- Keep the autoinjector in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Keep the autoinjector in the original carton to protect it from light or physical damage.
- Do not freeze the autoinjector.
- Do not store the autoinjector in extreme heat or cold. For example, avoid storing in your vehicle's glove box or trunk.
| Important: Keep the autoinjector out of the sight and reach of children. |
| WAIT |
2b Wait 15 to 30 minutes for the autoinjector to reach room temperature.
- Remove the number of autoinjectors you need for your injection and put any unused autoinjectors back into the refrigerator.
- Let the autoinjector warm up naturally.
- Do not heat the autoinjector with hot water, a microwave, or direct sunlight.
- Do not shake the autoinjector at any time.
- Using the autoinjector at room temperature makes sure the full dose is delivered and allows for a more comfortable injection.
2c You may keep AMJEVITA at room temperature for up to 14 days, if needed.
- For example, when you are traveling, you may keep AMJEVITA at room temperature.
- Keep it at room temperature between 68°F to 77°F (20°C to 25°C).
- Do not put it back in the refrigerator.
- Record the date you removed it from the refrigerator and use it within 14 days.
| Important: Place the autoinjector in a sharps disposal container if it has reached room temperature and has not been used within 14 days. |
2d Inspect the medicine. It should be clear and colorless to pale yellow.
- It is okay to see air bubbles in the autoinjector.
- Do not use AMJEVITA if the medicine is cloudy, discolored, or has flakes or particles.
| Important: If the medicine is cloudy, discolored, or has flakes or particles, or if the autoinjector is damaged or expired, call 1-800-77-AMGEN (1-800-772-6436). |
2e Check the expiration date (Exp.) and inspect the autoinjector for damage.
- Do not use the autoinjector if the expiration date has passed.
-
Do not use the autoinjector if:
- the yellow cap is missing or loose.
- it has cracks or broken parts.
- it has been dropped on a hard surface.
- Make sure you have the right medicine and dose.
3. Getting Ready for Your Injection
3a Gather and place the following items for your injection on a clean, flat, and well-lit surface:
- AMJEVITA autoinjector (room temperature)
- Sharps disposal container [see Disposing of AMJEVITA and Checking the Injection Site]
- Alcohol wipe
- Adhesive bandage
- Cotton ball or gauze pad
3b Inject into 1 of these locations.
- Inject into the front of your thigh or stomach (except for 2 inches around your belly button).
- Choose a different site for each injection.
| Important: Avoid areas with scars or stretch marks, or where the skin is tender, bruised, red, or hard. |
3c Wash hands thoroughly with soap and water.
3d Clean the injection site with an alcohol wipe.
- Let the skin dry on its own.
- Do not touch this area again before injecting.
4. Injecting AMJEVITA
| Important: Only remove the yellow cap when you can inject right away (within 5 minutes) because the medicine can dry out. Do not recap. |
4a Grasp the autoinjector so you can see the window. Pull the yellow cap straight off. You may need to pull hard.
- Do not twist, bend or wiggle the yellow cap to pull it off.
- Never put the yellow cap back on. It may damage the needle.
- Do not put your finger inside the cream safety guard.
- It is normal to see a drop of medicine at the end of the needle or cream safety guard.
4b Pinch the skin to create a firm surface at the injection site. Place the cream safety guard straight against the pinched skin.
- Keep the skin pinched until the injection is finished.
- Make sure you can see the window.
- Make sure the autoinjector is positioned straight on the injection site (at a 90-degree angle).
|
PUSH and hold against skin |
4c Firmly push the autoinjector down until the cream safety guard stops moving. Hold the autoinjector down, do not lift.
- The cream safety guard pushes in and unlocks the light blue start button.
|
PRESS light blue start button |
4d Keep pushing the autoinjector down and press the light blue start button to start the injection.
- You may hear or feel a click.
- The window starts to turn yellow.
- It is okay to let go of the light blue start button.
|
WATCH and CONFIRM that the window turns fully yellow |
4e Keep pushing the autoinjector down. When the window is fully yellow, the injection is complete.
- The injection may take up to 10 seconds to complete.
- You may hear or feel a click.
- Lift the autoinjector away from your skin.
- The cream safety guard locks around the needle.
| Important: If the window has not turned fully yellow, or it looks like the medicine is still coming out, you have not received a full dose. Call your healthcare provider right away. |
5. Disposing of AMJEVITA and Checking the Injection Site
5a Place the used autoinjector and yellow cap in an FDA-cleared sharps disposal container right away after use.
| Important: Do not throw away the autoinjector in your household trash. |
- Do not reuse the autoinjector.
- Do not touch the cream safety guard.
5b Check the injection site.
- Do not rub the injection site.
- If there is blood, press a cotton ball or gauze pad on your injection site. Apply an adhesive bandage if necessary.
Additional information about your sharps disposal container
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
Disposing of sharps disposal containers:
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container.
There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at:
http://www.fda.gov/safesharpsdisposal
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
Do not recycle your used sharps disposal container.
For more information or help call 1-800-77-AMGEN (1-800-772-6436).
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
AMJEVITA (adalimumab-atto)
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
US License Number 1080
©2024 Amgen Inc. All rights reserved.
1xxxxxx
Revised: 8/2024
v2
12.2 Pharmacodynamics
After treatment with adalimumab, a decrease in concentrations of acute phase reactants of inflammation (C-reactive protein [CRP] and erythrocyte sedimentation rate [ESR]) and serum cytokines (IL-6) was observed compared to baseline in patients with rheumatoid arthritis. A decrease in CRP concentrations was also observed in patients with Crohn's disease, ulcerative colitis, and hidradenitis suppurativa. Serum concentrations of matrix metalloproteinases (MMP-1 and MMP-3) that produce tissue remodeling responsible for cartilage destruction were also decreased after adalimumab administration.
12.3 Pharmacokinetics
The pharmacokinetics of adalimumab were linear over the dose range of 0.5 to 10 mg/kg following administration of a single intravenous dose (adalimumab products are not approved for intravenous use). Following 20, 40, and 80 mg every other week and every week subcutaneous administration, adalimumab mean serum trough concentrations at steady state increased approximately proportionally with dose in RA patients. The mean terminal half-life was approximately 2 weeks, ranging from 10 to 20 days across studies. Healthy subjects and patients with RA displayed similar adalimumab pharmacokinetics.
Adalimumab exposure in patients treated with 80 mg every other week is estimated to be comparable with that in patients treated with 40 mg every week.
1.6 Ulcerative Colitis
AMJEVITA is indicated for the treatment of moderately to severely active ulcerative colitis in adult patients.
5.1 Serious Infections
Patients treated with adalimumab products, including AMJEVITA, are at increased risk for developing serious infections involving various organ systems and sites that may lead to hospitalization or death. Opportunistic infections due to bacterial, mycobacterial, invasive fungal, viral, parasitic, or other opportunistic pathogens including aspergillosis, blastomycosis, candidiasis, coccidioidomycosis, histoplasmosis, legionellosis, listeriosis, pneumocystosis and tuberculosis have been reported with TNF blockers. Patients have frequently presented with disseminated rather than localized disease.
The concomitant use of a TNF blocker and abatacept or anakinra was associated with a higher risk of serious infections in patients with rheumatoid arthritis (RA); therefore, the concomitant use of AMJEVITA and these biologic products is not recommended in the treatment of patients with RA [see Warnings and Precautions (5.7, 5.11) and Drug Interactions (7.2)].
Treatment with AMJEVITA should not be initiated in patients with an active infection, including localized infections. Patients 65 years of age and older, patients with co-morbid conditions and/or patients taking concomitant immunosuppressants (such as corticosteroids or methotrexate), may be at greater risk of infection. Consider the risks and benefits of treatment prior to initiating therapy in patients:
- with chronic or recurrent infection;
- who have been exposed to tuberculosis;
- with a history of an opportunistic infection;
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses, such as histoplasmosis, coccidioidomycosis, or blastomycosis; or
- with underlying conditions that may predispose them to infection.
1 Indications and Usage (1 INDICATIONS AND USAGE)
AMJEVITA is a tumor necrosis factor (TNF) blocker indicated for:
- Reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis. (1.1)
- Reducing signs and symptoms of moderately to severely active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older. (1.2)
- Reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with active psoriatic arthritis. (1.3)
- Reducing signs and symptoms in adult patients with active ankylosing spondylitis. (1.4)
- Treatment of moderately to severely active Crohn's disease in adults and pediatric patients 6 years of age and older. (1.5)
- Treatment of moderately to severely active ulcerative colitis in adult patients. (1.6)
Limitations of Use: Effectiveness has not been established in patients who have lost response to or were intolerant to TNF blockers. - Treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy, and when other systemic therapies are medically less appropriate. (1.7)
- Treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older. (1.8)
- Treatment of non-infectious intermediate, posterior, and panuveitis in adults and pediatric patients 2 years of age and older. (1.9)
1.3 Psoriatic Arthritis
AMJEVITA is indicated for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with active psoriatic arthritis.
AMJEVITA can be used alone or in combination with non-biologic DMARDs.
7.2 Biological Products
In clinical studies in patients with RA, an increased risk of serious infections has been observed with the combination of TNF blockers with anakinra or abatacept, with no added benefit; therefore, use of AMJEVITA with abatacept or anakinra is not recommended in patients with RA [see Warnings and Precautions (5.7, 5.11)]. A higher rate of serious infections has also been observed in patients with RA treated with rituximab who received subsequent treatment with a TNF blocker. There is insufficient information regarding the concomitant use of AMJEVITA and other biologic products for the treatment of RA, PsA, AS, CD, UC, Ps, HS and UV. Concomitant administration of AMJEVITA with other biologic DMARDs (e.g., anakinra and abatacept) or other TNF blockers is not recommended based upon the possible increased risk for infections and other potential pharmacological interactions.
1.1 Rheumatoid Arthritis
AMJEVITA is indicated for reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis. AMJEVITA can be used alone or in combination with methotrexate or other non-biologic disease-modifying anti-rheumatic drugs (DMARDs).
12.1 Mechanism of Action
Adalimumab products bind specifically to TNF-alpha and block its interaction with the p55 and p75 cell surface TNF receptors. Adalimumab products also lyse surface TNF expressing cells in vitro in the presence of complement. Adalimumab products do not bind or inactivate lymphotoxin (TNF-beta). TNF is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Elevated concentrations of TNF are found in the synovial fluid of patients with RA, JIA, PsA, and AS and play an important role in both the pathologic inflammation and the joint destruction that are hallmarks of these diseases. Increased concentrations of TNF are also found in psoriasis plaques. In Ps, treatment with AMJEVITA may reduce the epidermal thickness and infiltration of inflammatory cells. The relationship between these pharmacodynamic activities and the mechanism(s) by which adalimumab products exert their clinical effects is unknown.
Adalimumab products also modulate biological responses that are induced or regulated by TNF, including changes in the concentrations of adhesion molecules responsible for leukocyte migration (ELAM-1, VCAM-1, and ICAM-1 with an IC50 of 1-2 × 10-10M).
5.5 Neurologic Reactions
Use of TNF blocking agents, including adalimumab products, has been associated with rare cases of new onset or exacerbation of clinical symptoms and/or radiographic evidence of central nervous system demyelinating disease, including multiple sclerosis (MS) and optic neuritis, and peripheral demyelinating disease, including Guillain-Barré syndrome. Exercise caution in considering the use of AMJEVITA in patients with preexisting or recent-onset central or peripheral nervous system demyelinating disorders; discontinuation of AMJEVITA should be considered if any of these disorders develop. There is a known association between intermediate uveitis and central demyelinating disorders.
1.4 Ankylosing Spondylitis
AMJEVITA is indicated for reducing signs and symptoms in adult patients with active ankylosing spondylitis.
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Serious infections: Do not start AMJEVITA during an active infection. If an infection develops, monitor carefully, and stop AMJEVITA if infection becomes serious. (5.1)
- Invasive fungal infections: For patients who develop a systemic illness on AMJEVITA, consider empiric antifungal therapy for those who reside or travel to regions where mycoses are endemic. (5.1)
- Malignancies: Incidence of malignancies was greater in adalimumab-treated patients than in controls. (5.2)
- Anaphylaxis or serious hypersensitivity reactions may occur. (5.3)
- Hepatitis B virus reactivation: Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop AMJEVITA and begin anti-viral therapy. (5.4)
- Demyelinating disease: Exacerbation or new onset, may occur. (5.5)
- Cytopenias, pancytopenia: Advise patients to seek immediate medical attention if symptoms develop, and consider stopping AMJEVITA. (5.6)
- Heart failure: Worsening or new onset, may occur. (5.8)
- Autoimmunity: Stop AMJEVITA if lupus-like syndrome or autoimmune hepatitis develop. (5.9)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
- Administer by subcutaneous injection (2)
Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis (2.2): - Adults: 40 mg every other week.
Some patients with RA not receiving methotrexate may benefit from increasing the dosage to 40 mg every week or 80 mg every other week.
Juvenile Idiopathic Arthritis or Pediatric Uveitis (2.3):
| Pediatric Weight 2 Years of Age and Older |
Recommended Dosage |
|---|---|
| 10 kg (22 lbs) to less than 15 kg (33 lbs) | 10 mg every other week |
| 15 kg (33 lbs) to less than 30 kg (66 lbs) | 20 mg every other week |
| 30 kg (66 lbs) and greater | 40 mg every other week |
Crohn's Disease (2.4):
- Adults: 160 mg on Day 1 (given in one day or split over two consecutive days); 80 mg on Day 15; and 40 mg every other week starting on Day 29.
-
Pediatric Patients 6 Years of Age and Older:
Pediatric Weight Recommended Dosage Days 1 and 15 Starting on Day 29 17 kg (37 lbs) to less than 40 kg (88 lbs) Day 1: 80 mg
Day 15: 40 mg20 mg every other week 40 kg (88 lbs) and greater Day 1: 160 mg (single-dose or split over two consecutive days)
Day 15: 80 mg40 mg every other week
Ulcerative Colitis (2.5):
- Adults: 160 mg on Day 1 (given in one day or split over two consecutive days), 80 mg on Day 15 and 40 mg every other week starting on Day 29. Discontinue in patients without evidence of clinical remission by eight weeks (Day 57).
Plaque Psoriasis or Adult Uveitis (2.6):
- Adults: 80 mg initial dose, followed by 40 mg every other week starting one week after initial dose.
Hidradenitis Suppurativa (2.7)
-
Adults:
- Day 1:160 mg (given in one day or split over two consecutive days)
- Day 15: 80 mg
- Day 29 and subsequent doses: 40 mg every week or 80 mg every other week
-
Adolescents 12 years of age and older:
Adolescent Weight Recommended Dosage 30 kg (66 lbs) to less than 60 kg (132 lbs) Day 1: 80 mg
Day 8 and subsequent doses: 40 mg every other week60 kg (132 lbs) and greater Day 1: 160 mg (given in one day or split over two consecutive days)
Day 15: 80 mg
Day 29 and subsequent doses: 40 mg every week or 80 mg every other week
5.6 Hematological Reactions
Rare reports of pancytopenia including aplastic anemia have been reported with TNF blocking agents. Adverse reactions of the hematologic system, including medically significant cytopenia (e.g., thrombocytopenia, leukopenia) have been infrequently reported with adalimumab products. The causal relationship of these reports to adalimumab products remains unclear. Advise all patients to seek immediate medical attention if they develop signs and symptoms suggestive of blood dyscrasias or infection (e.g., persistent fever, bruising, bleeding, pallor) while on AMJEVITA. Consider discontinuation of AMJEVITA therapy in patients with confirmed significant hematologic abnormalities.
1.8 Hidradenitis Suppurativa
AMJEVITA is indicated for the treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older.
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
AMJEVITA is a clear, colorless to slightly yellow solution available as:
-
Prefilled SureClick Autoinjector
-
Injection: 80 mg/0.8 mL in a single-dose prefilled SureClick autoinjector.
-
Injection: 40 mg/0.8 mL in a single-dose prefilled SureClick autoinjector.
-
Injection: 40 mg/0.4 mL in a single-dose prefilled SureClick autoinjector.
-
-
Prefilled Syringe
-
Injection: 80 mg/0.8 mL in a single-dose prefilled glass syringe.
-
Injection: 40 mg/0.8 mL in a single-dose prefilled glass syringe.
-
Injection: 40 mg/0.4 mL in a single-dose prefilled glass syringe.
-
Injection: 20 mg/0.4 mL in a single-dose prefilled glass syringe.
-
Injection: 20 mg/0.2 mL in a single-dose prefilled glass syringe.
-
Injection: 10 mg/0.2 mL in a single-dose prefilled glass syringe.
-
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of adalimumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to adalimumab products exposure.
Gastrointestinal disorders: Diverticulitis, large bowel perforations including perforations associated with diverticulitis and appendiceal perforations associated with appendicitis, pancreatitis
General disorders and administration site conditions: Pyrexia
Hepato-biliary disorders: Liver failure, hepatitis, autoimmune hepatitis
Immune system disorders: Sarcoidosis
Neoplasms benign, malignant and unspecified (including cysts and polyps): Merkel Cell Carcinoma (neuroendocrine carcinoma of the skin)
Nervous system disorders: Demyelinating disorders (e.g., optic neuritis, Guillain-Barré syndrome), cerebrovascular accident
Respiratory disorders: Interstitial lung disease, including pulmonary fibrosis, pulmonary embolism
Skin reactions: Stevens Johnson Syndrome, cutaneous vasculitis, erythema multiforme, new or worsening psoriasis (all sub-types including pustular and palmoplantar), alopecia, lichenoid skin reaction
Vascular disorders: Systemic vasculitis, deep vein thrombosis
5.3 Hypersensitivity Reactions
Anaphylaxis and angioneurotic edema have been reported following administration of adalimumab products. If an anaphylactic or other serious allergic reaction occurs, immediately discontinue administration of AMJEVITA and institute appropriate therapy. In clinical trials of adalimumab, hypersensitivity reactions (e.g., rash, anaphylactoid reaction, fixed drug reaction, non-specified drug reaction, urticaria) have been observed.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reaction with adalimumab was injection site reactions. In placebo-controlled trials, 20% of subjects treated with adalimumab developed injection site reactions (erythema and/or itching, hemorrhage, pain or swelling), compared to 14% of subjects receiving placebo. Most injection site reactions were described as mild and generally did not necessitate drug discontinuation.
The proportion of subjects who discontinued treatment due to adverse reactions during the double-blind, placebo-controlled portion of studies in subjects with RA (i.e., Studies RA-I, RA-II, RA-III and RA-IV) was 7% for subjects taking adalimumab and 4% for placebo-treated subjects. The most common adverse reactions leading to discontinuation of adalimumab in these RA studies were clinical flare reaction (0.7%), rash (0.3%) and pneumonia (0.3%).
7.4 Cytochrome P450 Substrates
The formation of CYP450 enzymes may be suppressed by increased concentrations of cytokines (e.g., TNFα, IL-6) during chronic inflammation. It is possible for products that antagonize cytokine activity, such as adalimumab products, to influence the formation of CYP450 enzymes. Upon initiation or discontinuation of AMJEVITA in patients being treated with CYP450 substrates with a narrow therapeutic index, monitoring of the effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) is recommended and the individual dose of the drug product may be adjusted as needed.
1.2 Juvenile Idiopathic Arthritis
AMJEVITA is indicated for reducing signs and symptoms of moderately to severely active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older. AMJEVITA can be used alone or in combination with methotrexate.
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
5.4 Hepatitis B Virus Reactivation
Use of TNF blockers, including AMJEVITA, may increase the risk of reactivation of hepatitis B virus (HBV) in patients who are chronic carriers of this virus. In some instances, HBV reactivation occurring in conjunction with TNF blocker therapy has been fatal. The majority of these reports have occurred in patients concomitantly receiving other medications that suppress the immune system, which may also contribute to HBV reactivation. Evaluate patients at risk for HBV infection for prior evidence of HBV infection before initiating TNF blocker therapy. Exercise caution in prescribing TNF blockers for patients identified as carriers of HBV. Adequate data are not available on the safety or efficacy of treating patients who are carriers of HBV with anti-viral therapy in conjunction with TNF blocker therapy to prevent HBV reactivation. For patients who are carriers of HBV and require treatment with TNF blockers, closely monitor such patients for clinical and laboratory signs of active HBV infection throughout therapy and for several months following termination of therapy. In patients who develop HBV reactivation, stop AMJEVITA and initiate effective anti-viral therapy with appropriate supportive treatment. The safety of resuming TNF blocker therapy after HBV reactivation is controlled is not known. Therefore, exercise caution when considering resumption of AMJEVITA therapy in this situation and monitor patients closely.
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
AMJEVITA® (adalimumab-atto) injection is supplied as a preservative-free, sterile, clear, colorless to slightly yellow solution for subcutaneous administration. AMJEVITA is supplied in a single-dose prefilled syringe (PFS) or single-dose prefilled SureClick autoinjector (AI). The AMJEVITA prefilled syringe and prefilled SureClick autoinjector are not made with natural rubber latex.
The following packaging configurations are available.
| Presentation | Number of Units/Pack | NDC number |
|---|---|---|
| 10 mg/0.2 mL prefilled glass syringe with a fixed 29 gauge needle | 1 | 55513-413-01 |
| 20 mg/0.2 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-399-01 |
| 72511-399-01 | ||
| 20 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-411-01 |
| 40 mg/0.4 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-479-01 |
| 2 | 55513-479-02 | |
| 72511-479-02 | ||
| 40 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-410-01 |
| 2 | 55513-410-02 | |
| 80 mg/0.8 mL prefilled syringe with a fixed 29 gauge needle | 1 | 55513-480-01 |
| 2 | 55513-480-02 | |
| 40 mg/0.4 mL Prefilled SureClick Autoinjector | 1 | 55513-482-01 72511-482-01 |
| 2 | 55513-482-02 72511-482-02 |
|
| 40 mg/0.8 mL Prefilled SureClick Autoinjector | 1 | 55513-400-01 72511-400-01 |
| 2 | 55513-400-02 72511-400-02 |
|
| 80 mg/0.8 mL Prefilled SureClick Autoinjector | 1 | 55513-481-01 |
| 2 | 55513-481-02 | |
| 72511-481-02 |
2.1 Recommended Tuberculosis Evaluation
Prior to initiating AMJEVITA and periodically during therapy, evaluate patients for active tuberculosis and test for latent infection [see Warnings and Precautions (5.1)].
14.9 Clinical Studies in Plaque Psoriasis
The safety and efficacy of adalimumab were assessed in randomized, double-blind, placebo-controlled studies in 1696 adult subjects with moderate to severe chronic plaque psoriasis (Ps) who were candidates for systemic therapy or phototherapy.
Study Ps-I evaluated 1212 subjects with chronic Ps with ≥ 10% body surface area (BSA) involvement, Physician's Global Assessment (PGA) of at least moderate disease severity, and Psoriasis Area and Severity Index (PASI) ≥ 12 within three treatment periods. In period A, subjects received placebo or adalimumab at an initial dose of 80 mg at Week 0 followed by a dose of 40 mg every other week starting at Week 1. After 16 weeks of therapy, subjects who achieved at least a PASI 75 response at Week 16, defined as a PASI score improvement of at least 75% relative to baseline, entered period B and received open-label 40 mg adalimumab every other week. After 17 weeks of open-label therapy, subjects who maintained at least a PASI 75 response at Week 33 and were originally randomized to active therapy in period A were re-randomized in period C to receive 40 mg adalimumab every other week or placebo for an additional 19 weeks. Across all treatment groups the mean baseline PASI score was 19 and the baseline Physician's Global Assessment score ranged from "moderate" (53%) to "severe" (41%) to "very severe" (6%).
Study Ps-II evaluated 99 subjects randomized to adalimumab and 48 subjects randomized to placebo with chronic plaque psoriasis with ≥ 10% BSA involvement and PASI ≥ 12. Subjects received placebo, or an initial dose of 80 mg adalimumab at Week 0 followed by 40 mg every other week starting at Week 1 for 16 weeks. Across all treatment groups the mean baseline PASI score was 21 and the baseline PGA score ranged from "moderate" (41%) to "severe" (51%) to "very severe" (8%).
Studies Ps-I and II evaluated the proportion of subjects who achieved "clear" or "minimal" disease on the 6-point PGA scale and the proportion of subjects who achieved a reduction in PASI score of at least 75% (PASI 75) from baseline at Week 16 (see Table 16 and 17).
Additionally, Study Ps-I evaluated the proportion of subjects who maintained a PGA of "clear" or "minimal" disease or a PASI 75 response after Week 33 and on or before Week 52.
| Adalimumab 40 mg every other week | Placebo | |
|---|---|---|
| N = 814 | N = 398 | |
| PGA: Clear or minimal Clear = no plaque elevation, no scale, plus or minus hyperpigmentation or diffuse pink or red coloration
Minimal = possible but difficult to ascertain whether there is slight elevation of plaque above normal skin, plus or minus surface dryness with some white coloration, plus or minus up to red coloration |
506 (62%) | 17 (4%) |
| PASI 75 | 578 (71%) | 26 (7%) |
| Adalimumab 40 mg every other week | Placebo | |
|---|---|---|
| N = 99 | N = 48 | |
| PGA: Clear or minimal Clear = no plaque elevation, no scale, plus or minus hyperpigmentation or diffuse pink or red coloration
Minimal = possible but difficult to ascertain whether there is slight elevation of plaque above normal skin, plus or minus surface dryness with some white coloration, plus or minus up to red coloration |
70 (71%) | 5 (10%) |
| PASI 75 | 77 (78%) | 9 (19%) |
Additionally, in Study Ps-I, subjects on adalimumab who maintained a PASI 75 were re-randomized to adalimumab (N = 250) or placebo (N = 240) at Week 33. After 52 weeks of treatment with adalimumab, more subjects on adalimumab maintained efficacy when compared to subjects who were re-randomized to placebo based on maintenance of PGA of "clear" or "minimal" disease (68% vs. 28%) or a PASI 75 (79% vs. 43%).
A total of 347 stable responders participated in a withdrawal and retreatment evaluation in an open-label extension study. Median time to relapse (decline to PGA "moderate" or worse) was approximately 5 months. During the withdrawal period, no subject experienced transformation to either pustular or erythrodermic psoriasis. A total of 178 subjects who relapsed re-initiated treatment with 80 mg of adalimumab, then 40 mg every other week beginning at Week 1. At Week 16, 69% (123/178) of subjects had a response of PGA "clear" or "minimal".
A randomized, double-blind study (Study Ps-III) compared the efficacy and safety of adalimumab versus placebo in 217 adult subjects. Subjects in the study had to have chronic plaque psoriasis of at least moderate severity on the PGA scale, fingernail involvement of at least moderate severity on a 5-point Physician's Global Assessment of Fingernail Psoriasis (PGA-F) scale, a Modified Nail Psoriasis Severity Index (mNAPSI) score for the target-fingernail of ≥ 8, and either a BSA involvement of at least 10% or a BSA involvement of at least 5% with a total mNAPSI score for all fingernails of ≥ 20. Subjects received an initial dose of 80 mg adalimumab followed by 40 mg every other week (starting one week after the initial dose) or placebo for 26 weeks followed by open-label adalimumab treatment for an additional 26 weeks. This study evaluated the proportion of subjects who achieved "clear" or "minimal" assessment with at least a 2-grade improvement on the PGA-F scale and the proportion of subjects who achieved at least a 75% improvement from baseline in the mNAPSI score (mNAPSI 75) at Week 26.
At Week 26, a higher proportion of subjects in the adalimumab group than in the placebo group achieved the PGA-F endpoint. Furthermore, a higher proportion of subjects in the adalimumab group than in the placebo group achieved mNAPSI 75 at Week 26 (see Table 18).
| Endpoint | Adalimumab 40 mg every other week Subjects received 80 mg of adalimumab at Week 0, followed by 40 mg every other week starting at Week 1.
N = 109 |
Placebo N = 108 |
|---|---|---|
| PGA-F: ≥ 2-grade improvement and clear or minimal | 49% | 7% |
| mNAPSI 75 | 47% | 3% |
Nail pain was also evaluated and improvement in nail pain was observed in Study Ps-III.
Warning: Serious Infections and Malignancy (WARNING: SERIOUS INFECTIONS AND MALIGNANCY)
WARNING: SERIOUS INFECTIONS AND MALIGNANCY
See full prescribing information for complete boxed warning.
SERIOUS INFECTIONS (5.1, 6.1):
- Increased risk of serious infections leading to hospitalization or death, including tuberculosis (TB), bacterial sepsis, invasive fungal infections (such as histoplasmosis), and infections due to other opportunistic pathogens.
- Discontinue AMJEVITA if a patient develops a serious infection or sepsis during treatment.
- Perform test for latent TB; if positive, start treatment for TB prior to starting AMJEVITA.
- Monitor all patients for active TB during treatment, even if initial latent TB test is negative.
MALIGNANCY (5.2):
- Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers including adalimumab products.
- Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have occurred in adolescent and young adults with inflammatory bowel disease treated with TNF blockers including adalimumab products.
14.3 Clinical Studies in Psoriatic Arthritis
The safety and efficacy of adalimumab was assessed in two randomized, double-blind, placebo-controlled studies in 413 subjects with psoriatic arthritis (PsA). Upon completion of both studies, 383 subjects enrolled in an open-label extension study, in which 40 mg adalimumab was administered every other week.
Study PsA-I enrolled 313 adult subjects with moderately to severely active PsA (> 3 swollen and > 3 tender joints) who had an inadequate response to NSAID therapy in one of the following forms: (1) distal interphalangeal (DIP) involvement (N = 23); (2) polyarticular arthritis (absence of rheumatoid nodules and presence of plaque psoriasis) (N = 210); (3) arthritis mutilans (N = 1); (4) asymmetric PsA (N = 77); or (5) AS-like (N = 2). Subjects on MTX therapy (158 of 313 subjects) at enrollment (stable dose of ≤ 30 mg/week for > 1 month) could continue MTX at the same dose. Doses of adalimumab 40 mg or placebo every other week were administered during the 24-week double-blind period of the study.
Compared to placebo, treatment with adalimumab resulted in improvements in the measures of disease activity (see Tables 8 and 9). Among subjects with PsA who received adalimumab, the clinical responses were apparent in some subjects at the time of the first visit (two weeks) and were maintained up to 88 weeks in the ongoing open-label study. Similar responses were seen in subjects with each of the subtypes of psoriatic arthritis, although few subjects were enrolled with the arthritis mutilans and ankylosing spondylitis-like subtypes. Responses were similar in subjects who were or were not receiving concomitant MTX therapy at baseline.
Subjects with psoriatic involvement of at least three percent body surface area (BSA) were evaluated for Psoriatic Area and Severity Index (PASI) responses. At 24 weeks, the proportions of subjects achieving a 75% or 90% improvement in the PASI were 59% and 42%, respectively, in the adalimumab group (N = 69), compared to 1% and 0%, respectively, in the placebo group (N = 69) (p < 0.001). PASI responses were apparent in some subjects at the time of the first visit (two weeks). Responses were similar in subjects who were or were not receiving concomitant MTX therapy at baseline.
| Placebo N = 162 |
Adalimumab p < 0.001 for all comparisons between adalimumab and placebo
N = 151 |
|
|---|---|---|
| ACR 20 | ||
| Week 12 | 14% | 58% |
| Week 24 | 15% | 57% |
| ACR 50 | ||
| Week 12 | 4% | 36% |
| Week 24 | 6% | 39% |
| ACR 70 | ||
| Week 12 | 1% | 20% |
| Week 24 | 1% | 23% |
| Placebo N = 162 |
Adalimumab p < 0.001 for adalimumab vs. placebo comparisons based on median changes
N = 151 |
|||
|---|---|---|---|---|
| Parameter: median | Baseline | 24 weeks | Baseline | 24 weeks |
| Number of tender joints Scale 0-78
|
23.0 | 17.0 | 20.0 | 5.0 |
| Number of swollen joints Scale 0-76
|
11.0 | 9.0 | 11.0 | 3.0 |
| Physician global assessment Visual analog scale; 0 = best, 100 = worst
|
53.0 | 49.0 | 55.0 | 16.0 |
| Patient global assessment | 49.5 | 49.0 | 48.0 | 20.0 |
| Pain | 49.0 | 49.0 | 54.0 | 20.0 |
| Disability index (HAQ) Disability Index of the Health Assessment Questionnaire; 0 = best, 3 = worst; measures the patient's ability to perform the following: dress/groom, arise, eat, walk, reach, grip, maintain hygiene, and maintain daily activity
|
1.0 | 0.9 | 1.0 | 0.4 |
| CRP (mg/dL) Normal range: 0-0.287 mg/dL
|
0.8 | 0.7 | 0.8 | 0.2 |
Similar results were seen in an additional, 12-week study in 100 subjects with moderate to severe psoriatic arthritis who had suboptimal response to DMARD therapy as manifested by ≥ 3 tender joints and ≥ 3 swollen joints at enrollment.
14.1 Clinical Studies in Rheumatoid Arthritis
The efficacy and safety of adalimumab were assessed in five randomized, double-blind studies in subjects ≥ 18 years of age with active rheumatoid arthritis (RA) diagnosed according to American College of Rheumatology (ACR) criteria. Subjects had at least 6 swollen and 9 tender joints. Adalimumab was administered subcutaneously in combination with methotrexate (MTX) (12.5 to 25 mg, Studies RA-I, RA-III and RA-V) or as monotherapy (Studies RA-II and RA-V) or with other disease-modifying anti-rheumatic drugs (DMARDs) (Study RA-IV).
Study RA-I evaluated 271 subjects who had failed therapy with at least one but no more than four DMARDs and had inadequate response to MTX. Doses of 20, 40 or 80 mg of adalimumab or placebo were given every other week for 24 weeks.
Study RA-II evaluated 544 subjects who had failed therapy with at least one DMARD. Doses of placebo, 20 or 40 mg of adalimumab were given as monotherapy every other week or weekly for 26 weeks.
Study RA-III evaluated 619 subjects who had an inadequate response to MTX. Subjects received placebo, 40 mg of adalimumab every other week with placebo injections on alternate weeks, or 20 mg of adalimumab weekly for up to 52 weeks. Study RA-III had an additional primary endpoint at 52 weeks of inhibition of disease progression (as detected by X-ray results). Upon completion of the first 52 weeks, 457 subjects enrolled in an open-label extension phase in which 40 mg of adalimumab was administered every other week for up to 5 years.
Study RA-IV assessed safety in 636 subjects who were either DMARD-naive or were permitted to remain on their pre-existing rheumatologic therapy provided that therapy was stable for a minimum of 28 days. Subjects were randomized to 40 mg of adalimumab or placebo every other week for 24 weeks.
Study RA-V evaluated 799 subjects with moderately to severely active RA of less than 3 years duration who were ≥ 18 years old and MTX naïve. Subjects were randomized to receive either MTX (optimized to 20 mg/week by Week 8), adalimumab 40 mg every other week or adalimumab/MTX combination therapy for 104 weeks. Subjects were evaluated for signs and symptoms, and for radiographic progression of joint damage. The median disease duration among subjects enrolled in the study was 5 months. The median MTX dose achieved was 20 mg.
14.11 Clinical Studies in Adults With Uveitis (14.11 Clinical Studies in Adults with Uveitis)
The safety and efficacy of adalimumab were assessed in adult subjects with non-infectious intermediate, posterior and panuveitis excluding subjects with isolated anterior uveitis, in two randomized, double-masked, placebo-controlled studies (UV I and II). Subjects received placebo or adalimumab at an initial dose of 80 mg followed by 40 mg every other week starting one week after the initial dose. The primary efficacy endpoint in both studies was ´time to treatment failure´.
Treatment failure was a multi-component outcome defined as the development of new inflammatory chorioretinal and/or inflammatory retinal vascular lesions, an increase in anterior chamber (AC) cell grade or vitreous haze (VH) grade or a decrease in best corrected visual acuity (BCVA).
Study UV I evaluated 217 subjects with active uveitis while being treated with corticosteroids (oral prednisone at a dose of 10 to 60 mg/day). All subjects received a standardized dose of prednisone 60 mg/day at study entry followed by a mandatory taper schedule, with complete corticosteroid discontinuation by Week 15.
Study UV II evaluated 226 subjects with inactive uveitis while being treated with corticosteroids (oral prednisone 10 to 35 mg/day) at baseline to control their disease. Subjects subsequently underwent a mandatory taper schedule, with complete corticosteroid discontinuation by Week 19.
2.8 General Considerations for Administration
AMJEVITA is intended for use under the guidance and supervision of a physician. A patient may self-inject AMJEVITA or a caregiver may inject AMJEVITA using either the AMJEVITA prefilled SureClick autoinjector or prefilled syringe if a physician determines that it is appropriate, and with medical follow-up, as necessary, after proper training in subcutaneous injection technique.
AMJEVITA can be taken out of the refrigerator for 15 to 30 minutes before injecting to allow the liquid to come to room temperature. Do not remove the cap or cover while allowing it to reach room temperature. Carefully inspect the solution in the AMJEVITA prefilled SureClick autoinjector or prefilled syringe for particulate matter and discoloration prior to subcutaneous administration. If particulates and discolorations are noted, do not use the product. AMJEVITA does not contain preservatives; therefore, discard unused portions of drug remaining from the syringe.
Instruct patients using the AMJEVITA prefilled SureClick autoinjector or prefilled syringe to inject the full amount in the syringe, according to the directions provided in the Instructions for Use [see Instructions for Use].
Injections should occur at separate sites in the thigh or abdomen. Rotate injection sites and do not give injections into areas where the skin is tender, bruised, red or hard.
If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
Principal Display Panel 20 Mg Syringe Carton (PRINCIPAL DISPLAY PANEL - 20 mg Syringe Carton)
20 mg/
0.4 mL
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-411-01
Not made with
natural rubber latex
For Use in Pediatric Patients
15 kg to < 30 kg
20 mg/0.4 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze.
Do not Shake. Protect from light.
(see side panel for additional storage information)
ATTENTION: Enclosed Medication Guide is required
for each patient.
Contains 1 Single-dose
Prefilled Syringe
CAUTION, Consult
Accompanying Documents
Do not re-use
Rx Only
AMGEN®
Principal Display Panel 40 Mg Syringe Carton (PRINCIPAL DISPLAY PANEL - 40 mg Syringe Carton)
40
mg
/0.8 mL
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-410-01
Not made with
natural rubber latex
40 mg/0.8 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze.
Do not Shake. Protect from light.
(see side panel for additional storage information)
ATTENTION: Enclosed Medication Guide is required
for each patient.
Contains 1 Single-dose
Prefilled Syringe
CAUTION, Consult
Accompanying Documents
Do not re-use
Rx Only
AMGEN®
Principal Display Panel 80 Mg Syringe Carton (PRINCIPAL DISPLAY PANEL - 80 mg Syringe Carton)
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-480-01
80 mg/
0.8 mL
80 mg/0.8 mL
For Subcutaneous Use Only.
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use. Do not Shake.
Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is
required for each patient.
Rx Only
Do not use
if carton seal is broken
Not made with
natural rubber latex
1
Single-dose
Prefilled Syringe
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
14.4 Clinical Studies in Ankylosing Spondylitis
The safety and efficacy of adalimumab 40 mg every other week was assessed in 315 adult subjects in a randomized, 24 week double-blind, placebo-controlled study in subjects with active ankylosing spondylitis (AS) who had an inadequate response to glucocorticoids, NSAIDs, analgesics, methotrexate or sulfasalazine. Active AS was defined as subjects who fulfilled at least two of the following three criteria: (1) a Bath AS disease activity index (BASDAI) score ≥ 4 cm, (2) a visual analog score (VAS) for total back pain ≥ 40 mm, and (3) morning stiffness ≥ 1 hour. The blinded period was followed by an open-label period during which subjects received adalimumab 40 mg every other week subcutaneously for up to an additional 28 weeks.
Improvement in measures of disease activity was first observed at Week 2 and maintained through 24 weeks as shown in Figure 2 and Table 11.
Responses of subjects with total spinal ankylosis (n = 11) were similar to those without total ankylosis.
At 12 weeks, the ASAS 20/50/70 responses were achieved by 58%, 38%, and 23%, respectively, of subjects receiving adalimumab, compared to 21%, 10%, and 5%, respectively, of subjects receiving placebo (p < 0.001). Similar responses were seen at Week 24 and were sustained in subjects receiving open-label adalimumab for up to 52 weeks.
A greater proportion of subjects treated with adalimumab (22%) achieved a low level of disease activity at 24 weeks (defined as a value < 20 [on a scale of 0 to 100 mm] in each of the four ASAS response parameters) compared to subjects treated with placebo (6%).
| Placebo N = 107 |
Adalimumab N = 208 |
|||
|---|---|---|---|---|
| Baseline mean | Week 24 mean | Baseline mean | Week 24 mean | |
| ASAS 20 Response Criteria Statistically significant for comparisons between adalimumab and placebo at Week 24
|
||||
| Patient's Global Assessment of Disease Activity Percent of subjects with at least a 20% and 10-unit improvement measured on a Visual Analog Scale (VAS) with 0 = "none" and 100 = "severe"
|
65 | 60 | 63 | 38 |
| Total back pain | 67 | 58 | 65 | 37 |
| Inflammation mean of questions 5 and 6 of BASDAI (defined in 'd')
|
6.7 | 5.6 | 6.7 | 3.6 |
| BASFI Bath Ankylosing Spondylitis Functional Index
|
56 | 51 | 52 | 34 |
| BASDAI Bath Ankylosing Spondylitis Disease Activity Index score
|
6.3 | 5.5 | 6.3 | 3.7 |
| BASMI Bath Ankylosing Spondylitis Metrology Index score
|
4.2 | 4.1 | 3.8 | 3.3 |
| Tragus to wall (cm) | 15.9 | 15.8 | 15.8 | 15.4 |
| Lumbar flexion (cm) | 4.1 | 4.0 | 4.2 | 4.4 |
| Cervical rotation (degrees) | 42.2 | 42.1 | 48.4 | 51.6 |
| Lumbar side flexion (cm) | 8.9 | 9.0 | 9.7 | 11.7 |
| Intermalleolar distance (cm) | 92.9 | 94.0 | 93.5 | 100.8 |
| CRP C-Reactive Protein (mg/dL)
|
2.2 | 2.0 | 1.8 | 0.6 |
A second randomized, multicenter, double-blind, placebo-controlled study of 82 subjects with ankylosing spondylitis showed similar results.
Subjects treated with adalimumab achieved improvement from baseline in the Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL) score (-3.6 vs. -1.1) and in the Short Form Health Survey (SF-36) Physical Component Summary (PCS) score (7.4 vs. 1.9) compared to placebo-treated subjects at Week 24.
14.10 Clinical Studies in Hidradenitis Suppurativa
Two randomized, double-blind, placebo-controlled studies (Studies HS-I and II) evaluated the safety and efficacy of adalimumab in a total of 633 adult subjects with moderate to severe hidradenitis suppurativa (HS) with Hurley Stage II or III disease and with at least 3 abscesses or inflammatory nodules. In both studies, subjects received placebo or adalimumab at an initial dose of 160 mg at Week 0, 80 mg at Week 2, and 40 mg every week starting at Week 4 and continued through Week 11. Subjects used topical antiseptic wash daily. Concomitant oral antibiotic use was allowed in Study HS-II.
Both studies evaluated Hidradenitis Suppurativa Clinical Response (HiSCR) at Week 12. HiSCR was defined as at least a 50% reduction in total abscess and inflammatory nodule count with no increase in abscess count and no increase in draining fistula count relative to baseline (see Table 19). Reduction in HS-related skin pain was assessed using a Numeric Rating Scale in subjects who entered the study with an initial baseline score of 3 or greater on a 11 point scale.
In both studies, a higher proportion of adalimumab- than placebo-treated subjects achieved HiSCR (see Table 19).
| HS Study I | HS Study II 19.3% of subjects in Study HS-II continued baseline oral antibiotic therapy during the study.
|
|||
|---|---|---|---|---|
| Placebo | Adalimumab 40 mg Weekly | Placebo | Adalimumab 40 mg Weekly | |
| Hidradenitis Suppurativa Clinical Response (HiSCR) | N = 154 40 (26%) |
N = 153 64 (42%) |
N=163 45 (28%) |
N=163 96 (59%) |
In both studies, from Week 12 to Week 35 (Period B), subjects who had received adalimumab were re-randomized to 1 of 3 treatment groups (adalimumab 40 mg every week, adalimumab 40 mg every other week, or placebo). Subjects who had been randomized to placebo were assigned to receive adalimumab 40 mg every week (Study HS-I) or placebo (Study HS-II).
During Period B, flare of HS, defined as ≥ 25% increase from baseline in abscesses and inflammatory nodule counts and with a minimum of 2 additional lesions, was documented in 22 (22%) of the 100 subjects who were withdrawn from adalimumab treatment following the primary efficacy timepoint in two studies.
Principal Display Panel 40 Mg Autoinjector Carton (PRINCIPAL DISPLAY PANEL - 40 mg Autoinjector Carton)
40 mg/
0.8 mL
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-400-01
SureClick®
Prefilled Autoinjector
Not made with
natural rubber latex
40 mg/0.8 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze.
Do not Shake. Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is
required for each patient.
Contains 1 Single-dose
Prefilled Autoinjector
CAUTION, Consult
Accompanying Documents
Do not re-use
Rx Only
AMGEN®
Principal Display Panel 80 Mg Autoinjector Carton (PRINCIPAL DISPLAY PANEL - 80 mg Autoinjector Carton)
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-481-01
80 mg/
0.8 mL
80 mg/0.8 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use.
Do not Shake. Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is
required for each patient.
Rx Only
Not made with
natural rubber latex
1
Single-dose Prefilled
SureClick® Autoinjector
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
14.5 Clinical Studies in Adults With Crohn's Disease (14.5 Clinical Studies in Adults with Crohn's Disease)
The safety and efficacy of multiple doses of adalimumab were assessed in adult subjects with moderately to severely active Crohn's disease, CD, (Crohn's Disease Activity Index (CDAI) ≥ 220 and ≤ 450) in randomized, double-blind, placebo-controlled studies. Concomitant stable doses of aminosalicylates, corticosteroids, and/or immunomodulatory agents were permitted, and 79% of subjects continued to receive at least one of these medications.
Induction of clinical remission (defined as CDAI < 150) was evaluated in two studies. In Study CD-I, 299 TNF-blocker naïve subjects were randomized to one of four treatment groups: the placebo group received placebo at Weeks 0 and 2, the 160/80 group received 160 mg adalimumab at Week 0 and 80 mg at Week 2, the 80/40 group received 80 mg at Week 0 and 40 mg at Week 2, and the 40/20 group received 40 mg at Week 0 and 20 mg at Week 2. Clinical results were assessed at Week 4.
In the second induction study, Study CD-II, 325 subjects who had lost response to, or were intolerant to, previous infliximab therapy were randomized to receive either 160 mg adalimumab at Week 0 and 80 mg at Week 2, or placebo at Weeks 0 and 2. Clinical results were assessed at Week 4.
Maintenance of clinical remission was evaluated in Study CD-III. In this study, 854 subjects with active disease received open-label adalimumab, 80 mg at Week 0 and 40 mg at Week 2. Subjects were then randomized at Week 4 to 40 mg adalimumab every other week, 40 mg adalimumab every week, or placebo. The total study duration was 56 weeks. Subjects in clinical response (decrease in CDAI ≥ 70) at Week 4 were stratified and analyzed separately from those not in clinical response at Week 4.
Principal Display Panel 10 Mg/0.2 Ml Syringe Carton (PRINCIPAL DISPLAY PANEL - 10 mg/0.2 mL Syringe Carton)
AMGEN®
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-413-01
10 mg/
0.2 mL
1
Single-dose
Prefilled Syringe
10 mg/0.2 mL
For Subcutaneous Use Only.
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use. Do not Shake.
Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is required
for each patient.
Rx Only
Do not use
if carton seal is broken
Not made with
natural rubber latex
For Use in Pediatric Patients
10 kg to < 15 kg
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
Principal Display Panel 20 Mg/0.2 Ml Syringe Carton (PRINCIPAL DISPLAY PANEL - 20 mg/0.2 mL Syringe Carton)
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-399-01
20 mg/
0.2 mL
20 mg/0.2 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use. Do not Shake.
Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is required
for each patient.
Rx Only
Do not use
if carton seal is broken
Not made with
natural rubber latex
1
Single-dose
Prefilled Syringe
For Use in Pediatric Patients
15 kg to < 30 kg
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
Principal Display Panel 40 Mg/0.4 Ml Syringe Carton (PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Syringe Carton)
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-479-01
40 mg/
0.4 mL
40 mg/0.4 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use. Do not Shake.
Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide is required
for each patient.
Rx Only
Do not use
if carton seal is broken
Not made with
natural rubber latex
1
Single-dose
Prefilled Syringe
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
14.2 Clinical Studies in Juvenile Idiopathic Arthritis
The safety and efficacy of adalimumab was assessed in two studies (Studies JIA-I and JIA-II) in subjects with active polyarticular juvenile idiopathic arthritis (JIA).
14.7 Clinical Studies in Adults With Ulcerative Colitis (14.7 Clinical Studies in Adults with Ulcerative Colitis)
The safety and efficacy of adalimumab were assessed in adult subjects with moderately to severely active ulcerative colitis (Mayo score 6 to 12 on a 12 point scale, with an endoscopy subscore of 2 to 3 on a scale of 0 to 3) despite concurrent or prior treatment with immunosuppressants such as corticosteroids, azathioprine, or 6-MP in two randomized, double-blind, placebo-controlled clinical studies (Studies UC-I and UC-II). Both studies enrolled TNF-blocker naïve subjects, but Study UC-II also allowed entry of subjects who lost response to or were intolerant to TNF-blockers. Forty percent (40%) of subjects enrolled in Study UC-II had previously used another TNF-blocker.
Concomitant stable doses of aminosalicylates and immunosuppressants were permitted. In Studies UC-I and II, subjects were receiving aminosalicylates (69%), corticosteroids (59%) and/or azathioprine or 6-MP (37%) at baseline. In both studies, 92% of subjects received at least one of these medications.
Induction of clinical remission (defined as Mayo score ≤ 2 with no individual subscores > 1) at Week 8 was evaluated in both studies. Clinical remission at Week 52 and sustained clinical remission (defined as clinical remission at both Weeks 8 and 52) were evaluated in Study UC-II.
In Study UC-I, 390 TNF-blocker naïve subjects were randomized to one of three treatment groups for the primary efficacy analysis. The placebo group received placebo at Weeks 0, 2, 4 and 6. The 160/80 group received 160 mg adalimumab at Week 0 and 80 mg at Week 2, and the 80/40 group received 80 mg adalimumab at Week 0 and 40 mg at Week 2. After Week 2, subjects in both adalimumab treatment groups received 40 mg every other week.
In Study UC-II, 518 subjects were randomized to receive either adalimumab 160 mg at Week 0, 80 mg at Week 2, and 40 mg every other week starting at Week 4 through Week 50, or placebo starting at Week 0 and every other week through Week 50. Corticosteroid taper was permitted starting at Week 8.
In both Studies UC-I and UC-II, a greater percentage of the subjects treated with 160/80 mg of adalimumab compared to subjects treated with placebo achieved induction of clinical remission. In Study UC-II, a greater percentage of the subjects treated with 160/80 mg of adalimumab compared to subjects treated with placebo achieved sustained clinical remission (clinical remission at both Weeks 8 and 52) (see Table 15).
| Study UC-I | Study UC-II | |||||
|---|---|---|---|---|---|---|
| Placebo N = 130 |
Adalimumab 160/80 mg N = 130 |
Treatment Difference (95% CI) |
Placebo N = 246 |
Adalimumab 160/80 mg N = 248 |
Treatment Difference (95% CI) |
|
| Clinical remission is defined as Mayo score ≤ 2 with no individual subscores > 1. CI = Confidence interval |
||||||
| Induction of Clinical Remission (Clinical Remission at Week 8) | 9.2% | 18.5% | 9.3% p < 0.05 for adalimumab vs. placebo pairwise comparison of proportions
(0.9%, 17.6%) |
9.3% | 16.5% | 7.2%
(1.2%, 12.9%) |
| Sustained Clinical Remission (Clinical Remission at both Weeks 8 and 52) | N/A | N/A | N/A | 4.1% | 8.5% | 4.4%
(0.1%, 8.6%) |
In Study UC-I, there was no statistically significant difference in clinical remission observed between the adalimumab 80/40 mg group and the placebo group at Week 8.
In Study UC-II, 17.3% (43/248) in the adalimumab group were in clinical remission at Week 52 compared to 8.5% (21/246) in the placebo group (treatment difference: 8.8%; 95% confidence interval (CI): [2.8%, 14.5%]; p < 0.05).
In the subgroup of subjects in Study UC-II with prior TNF-blocker use, the treatment difference for induction of clinical remission appeared to be lower than that seen in the whole study population, and the treatment differences for sustained clinical remission and clinical remission at Week 52 appeared to be similar to those seen in the whole study population. The subgroup of subjects with prior TNF-blocker use achieved induction of clinical remission at 9% (9/98) in the adalimumab group versus 7% (7/101) in the placebo group, and sustained clinical remission at 5% (5/98) in the adalimumab group versus 1% (1/101) in the placebo group. In the subgroup of subjects with prior TNF-blocker use, 10% (10/98) were in clinical remission at Week 52 in the adalimumab group versus 3% (3/101) in the placebo group.
5.7 Increased Risk of Infection When Used With Anakinra (5.7 Increased Risk of Infection When Used with Anakinra)
Concurrent use of anakinra (an interleukin-1 antagonist) and another TNF-blocker, was associated with a greater proportion of serious infections and neutropenia and no added benefit compared with the TNF-blocker alone in patients with RA. Therefore, the combination of AMJEVITA and anakinra is not recommended [see Drug Interactions (7.2)].
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies of adalimumab products have not been conducted to evaluate the carcinogenic potential or its effect on fertility.
14.12 Clinical Studies in Pediatric Subjects With Uveitis (14.12 Clinical Studies in Pediatric Subjects with Uveitis)
The safety and efficacy of adalimumab were assessed in a randomized, double-masked, placebo-controlled study of 90 pediatric subjects from 2 to < 18 years of age with active JIA-associated non-infectious uveitis (PUV-I). Subjects received either placebo or 20 mg adalimumab (if < 30 kg) or 40 mg adalimumab (if ≥ 30 kg) every other week in combination with a dose of methotrexate. Concomitant dosages of corticosteroids were permitted at study entry followed by a mandatory reduction in topical corticosteroids within 3 months.
The primary endpoint was 'time to treatment failure'. The criteria determining treatment failure were worsening or sustained non-improvement in ocular inflammation, or worsening of ocular co-morbidities.
5.11 Increased Risk of Infection When Used With Abatacept (5.11 Increased Risk of Infection When Used with Abatacept)
In controlled trials, the concurrent administration of TNF-blockers and abatacept was associated with a greater proportion of serious infections than the use of a TNF-blocker alone; the combination therapy, compared to the use of a TNF-blocker alone, has not demonstrated improved clinical benefit in the treatment of RA. Therefore, the combination of abatacept with TNF-blockers including AMJEVITA is not recommended [see Drug Interactions (7.2)].
Principal Display Panel 40 Mg/0.4 Ml Autoinjector Carton (PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Autoinjector Carton)
AMJEVITA™
(adalimumab-atto)
Injection
NDC 55513-482-01
40 mg/
0.4 mL
40 mg/0.4 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Do not Freeze. Do not re-use.
Do not Shake. Protect from light.
(see side panel for additional storage information)
Keep out of the sight and reach of children.
ATTENTION: Enclosed Medication Guide
is required for each patient.
Rx Only
Not made with
natural rubber latex
1
Single-dose Prefilled
SureClick® Autoinjector
Refrigerate
until ready
to use
CAUTION, Consult
Accompanying Documents
AMGEN®
14.6 Clinical Studies in Pediatric Subjects With Crohn's Disease (14.6 Clinical Studies in Pediatric Subjects with Crohn's Disease)
A randomized, double-blind, 52-week clinical study of 2 dose concentrations of adalimumab (Study PCD-I) was conducted in 192 pediatric subjects (6 to 17 years of age) with moderately to severely active Crohn's disease (defined as Pediatric Crohn's Disease Activity Index (PCDAI) score > 30). Enrolled subjects had over the previous two year period an inadequate response to corticosteroids or an immunomodulator (i.e., azathioprine, 6-mercaptopurine, or methotrexate). Subjects who had previously received a TNF blocker were allowed to enroll if they had previously had loss of response or intolerance to that TNF blocker.
Subjects received open-label induction therapy at a dose based on their body weight (≥ 40 kg and < 40 kg). Subjects weighing ≥ 40 kg received 160 mg (at Week 0) and 80 mg (at Week 2). Subjects weighing < 40 kg received 80 mg (at Week 0) and 40 mg (at Week 2). At Week 4, subjects within each body weight category (≥ 40 kg and < 40 kg) were randomized 1:1 to one of two maintenance dose regimens (high dose and low dose). The high dose was 40 mg every other week for subjects weighing ≥ 40 kg and 20 mg every other week for subjects weighing < 40 kg. The low dose was 20 mg every other week for subjects weighing ≥ 40 kg and 10 mg every other week for subjects weighing < 40 kg.
Concomitant stable dosages of corticosteroids (prednisone dosage ≤ 40 mg/day or equivalent) and immunomodulators (azathioprine, 6-mercaptopurine, or methotrexate) were permitted throughout the study.
At Week 12, subjects who experienced a disease flare (increase in PCDAI of ≥ 15 from Week 4 and absolute PCDAI > 30) or who were non-responders (did not achieve a decrease in the PCDAI of ≥ 15 from baseline for 2 consecutive visits at least 2 weeks apart) were allowed to dose-escalate (i.e., switch from blinded every other week dosing to blinded every week dosing); subjects who dose-escalated were considered treatment failures.
At baseline, 38% of subjects were receiving corticosteroids, and 62% of subjects were receiving an immunomodulator. Forty-four percent (44%) of subjects had previously lost response or were intolerant to a TNF blocker. The median baseline PCDAI score was 40.
Of the 192 subjects total, 188 subjects completed the 4 week induction period, 152 subjects completed 26 weeks of treatment, and 124 subjects completed 52 weeks of treatment. Fifty-one percent (51%) (48/95) of subjects in the low maintenance dose group dose-escalated, and 38% (35/93) of subjects in the high maintenance dose group dose-escalated.
At Week 4, 28% (52/188) of subjects were in clinical remission (defined as PCDAI ≤ 10).
The proportions of subjects in clinical remission (defined as PCDAI ≤ 10) and clinical response (defined as reduction in PCDAI of at least 15 points from baseline) were assessed at Weeks 26 and 52.
At both Weeks 26 and 52, the proportion of subjects in clinical remission and clinical response was numerically higher in the high dose group compared to the low dose group (Table 14). The recommended maintenance regimen is 20 mg every other week for subjects weighing < 40 kg and 40 mg every other week for subjects weighing ≥ 40 kg. Every week dosing is not the recommended maintenance dosing regimen [see Dosage and Administration (2.3)].
| Low Maintenance Dose The low maintenance dose was 20 mg every other week for subjects weighing ≥ 40 kg and 10 mg every other week for subjects weighing < 40 kg.
(20 or 10 mg every other week) N = 95 |
High Maintenance Dose The high maintenance dose was 40 mg every other week for subjects weighing ≥ 40 kg and 20 mg every other week for subjects weighing < 40 kg.
(40 or 20 mg every other week) N = 93 |
|
|---|---|---|
| Week 26 | ||
| Clinical remission Clinical remission defined as PCDAI ≤ 10.
|
28% | 39% |
| Clinical response Clinical response defined as reduction in PCDAI of at least 15 points from baseline.
|
48% | 59% |
| Week 52 | ||
| Clinical remission | 23% | 33% |
| Clinical response | 28% | 42% |
2.6 Recommended Dosage in Plaque Psoriasis Or Adults With Uveitis (2.6 Recommended Dosage in Plaque Psoriasis or Adults with Uveitis)
The recommended subcutaneous dosage of AMJEVITA for adult patients with plaque psoriasis (Ps) or uveitis (UV) [see Indications and Usage (1.7, 1.9)] is an initial dose of 80 mg, followed by 40 mg given every other week starting one week after the initial dose. The use of adalimumab products in moderate to severe chronic Ps beyond one year has not been evaluated in controlled clinical studies.
2.3 Recommended Dosage in Juvenile Idiopathic Arthritis Or Pediatric Patients With Uveitis (2.3 Recommended Dosage in Juvenile Idiopathic Arthritis or Pediatric Patients with Uveitis)
The recommended subcutaneous dosage of AMJEVITA for pediatric patients 2 years of age and older with polyarticular juvenile idiopathic arthritis (JIA) or pediatric uveitis [see Indications and Usage (1.2, 1.9)], based on weight, is shown below. MTX, glucocorticoids, NSAIDs, and/or analgesics may be continued during treatment with AMJEVITA.
| Pediatric Weight (2 Years of Age and Older) | Recommended Dosage |
|---|---|
| 10 kg (22 lbs) to less than 15 kg (33 lbs) | 10 mg every other week |
| 15 kg (33 lbs) to less than 30 kg (66 lbs) | 20 mg every other week |
| 30 kg (66 lbs) and greater | 40 mg every other week |
Adalimumab products have not been studied in patients with polyarticular JIA or pediatric uveitis less than 2 years of age or in patients with a weight below 10 kg.
2.2 Recommended Dosage in Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
The recommended subcutaneous dosage of AMJEVITA for adult patients with rheumatoid arthritis (RA), psoriatic arthritis (PsA), or ankylosing spondylitis (AS) [see Indication and Usage (1.1, 1.3, 1.4)] is 40 mg administered every other week. Methotrexate (MTX), other non-biologic DMARDs, glucocorticoids, nonsteroidal anti-inflammatory drugs (NSAIDs), and/or analgesics may be continued during treatment with AMJEVITA. In the treatment of RA, some patients not taking concomitant MTX may derive additional benefit from increasing the dosage of AMJEVITA to 40 mg every week or 80 mg every other week.
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:49:59.645577 · Updated: 2026-03-14T22:48:45.434128