Ryanodex
8f7b3ac0-604d-4c78-b545-5e0f8ea3d698
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
RYANODEX ® is indicated for the: Treatment of malignant hyperthermia in conjunction with appropriate supportive measures [see Dosage and Administration ( 2.1 )] Prevention of malignant hyperthermia in patients at high risk.
Dosage and Administration
Treatment of Malignant Hyperthermia (MH) ( 2.1 ) Administer by intravenous push at a minimum of 1 mg/kg. If signs continue, administer additional intravenous boluses up to maximum cumulative dosage of 10 mg/kg. Institute supportive measures (e.g., discontinue MH-triggering agents, manage metabolic acidosis, cooling if necessary, administer diuretics) Prevention of MH in Patients at High Risk ( 2.2 ) Administer 2.5 mg/kg intravenously over a period of at least 1 minute, starting approximately 75 minutes prior to surgery. Avoid agents that trigger MH. Administer additional individualized doses during anesthesia and surgery if surgery is prolonged. Pediatric Patients : recommended weight-based dose is the same as for adults ( 2.3 ) Reconstitution : With 5 mL of sterile water for injection (without a bacteriostatic agent) prior to administration ( 2.4 )
Contraindications
None
Warnings and Precautions
Skeletal Muscle Weakness : Ambulate patients with assistance until they have normal strength and balance ( 5.1 ) Dyspnea, Respiratory Muscle Weakness, and Decreased Inspiratory Capacity : Monitor patients for the adequacy of ventilation ( 5.1 ) Dysphagia : Assess patients for difficulty swallowing and choking ( 5.1 ) Somnolence and Dizziness : Can occur following RYANODEX administration; may persist up to 48 hours post-dose. Ambulate patients with assistance until they have normal strength and balance ( 5.2 ) Tissue Necrosis with Extravasation : Due to high pH of reconstituted RYANODEX for injectable suspension, care must be taken to prevent extravasation into surrounding tissues ( 5.3 )
Adverse Reactions
Administration to conscious subjects is associated with loss of grip strength and weakness in the legs, drowsiness and dizziness. Other common adverse reactions are: nausea, thrombophlebitis, tissue necrosis secondary to extravasation, urticaria and erythema, and injection site reactions (pain, erythema, swelling) ( 6.1 ) To report SUSPECTED ADVERSE REACTIONS, contact Eagle at 1-855-318-2170 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Calcium Channel Blockers : Concomitant use can cause cardiovascular collapse in association with marked hyperkalemia ( 7.1 ) Muscle Relaxants : Concomitant use with muscle relaxants may potentiate their effects ( 7.2 ) Sedatives : Concomitant use with sedative agents may potentiate their effects ( 7.3 )
How Supplied
RYANODEX ® (NDC 42367-540-32) is available in 20 mL vials containing a sterile lyophilized mixture of 250 mg dantrolene sodium for reconstitution with 5 mL sterile water for injection USP (without a bacteriostatic agent) to yield an orange colored injectable suspension. Store unreconstituted product at 20 °C to 25 °C (68 ºF to 77 ºF) [see USP Controlled Room Temperature], with excursions permitted to 15 ºC to 30 ºC (59 ºF to 86 ºF) and avoid prolonged exposure to light.
Storage and Handling
RYANODEX ® (NDC 42367-540-32) is available in 20 mL vials containing a sterile lyophilized mixture of 250 mg dantrolene sodium for reconstitution with 5 mL sterile water for injection USP (without a bacteriostatic agent) to yield an orange colored injectable suspension. Store unreconstituted product at 20 °C to 25 °C (68 ºF to 77 ºF) [see USP Controlled Room Temperature], with excursions permitted to 15 ºC to 30 ºC (59 ºF to 86 ºF) and avoid prolonged exposure to light.
Description
RYANODEX ® is indicated for the: Treatment of malignant hyperthermia in conjunction with appropriate supportive measures [see Dosage and Administration ( 2.1 )] Prevention of malignant hyperthermia in patients at high risk.
Medication Information
Warnings and Precautions
Skeletal Muscle Weakness : Ambulate patients with assistance until they have normal strength and balance ( 5.1 ) Dyspnea, Respiratory Muscle Weakness, and Decreased Inspiratory Capacity : Monitor patients for the adequacy of ventilation ( 5.1 ) Dysphagia : Assess patients for difficulty swallowing and choking ( 5.1 ) Somnolence and Dizziness : Can occur following RYANODEX administration; may persist up to 48 hours post-dose. Ambulate patients with assistance until they have normal strength and balance ( 5.2 ) Tissue Necrosis with Extravasation : Due to high pH of reconstituted RYANODEX for injectable suspension, care must be taken to prevent extravasation into surrounding tissues ( 5.3 )
Indications and Usage
RYANODEX ® is indicated for the: Treatment of malignant hyperthermia in conjunction with appropriate supportive measures [see Dosage and Administration ( 2.1 )] Prevention of malignant hyperthermia in patients at high risk.
Dosage and Administration
Treatment of Malignant Hyperthermia (MH) ( 2.1 ) Administer by intravenous push at a minimum of 1 mg/kg. If signs continue, administer additional intravenous boluses up to maximum cumulative dosage of 10 mg/kg. Institute supportive measures (e.g., discontinue MH-triggering agents, manage metabolic acidosis, cooling if necessary, administer diuretics) Prevention of MH in Patients at High Risk ( 2.2 ) Administer 2.5 mg/kg intravenously over a period of at least 1 minute, starting approximately 75 minutes prior to surgery. Avoid agents that trigger MH. Administer additional individualized doses during anesthesia and surgery if surgery is prolonged. Pediatric Patients : recommended weight-based dose is the same as for adults ( 2.3 ) Reconstitution : With 5 mL of sterile water for injection (without a bacteriostatic agent) prior to administration ( 2.4 )
Contraindications
None
Adverse Reactions
Administration to conscious subjects is associated with loss of grip strength and weakness in the legs, drowsiness and dizziness. Other common adverse reactions are: nausea, thrombophlebitis, tissue necrosis secondary to extravasation, urticaria and erythema, and injection site reactions (pain, erythema, swelling) ( 6.1 ) To report SUSPECTED ADVERSE REACTIONS, contact Eagle at 1-855-318-2170 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Calcium Channel Blockers : Concomitant use can cause cardiovascular collapse in association with marked hyperkalemia ( 7.1 ) Muscle Relaxants : Concomitant use with muscle relaxants may potentiate their effects ( 7.2 ) Sedatives : Concomitant use with sedative agents may potentiate their effects ( 7.3 )
Storage and Handling
RYANODEX ® (NDC 42367-540-32) is available in 20 mL vials containing a sterile lyophilized mixture of 250 mg dantrolene sodium for reconstitution with 5 mL sterile water for injection USP (without a bacteriostatic agent) to yield an orange colored injectable suspension. Store unreconstituted product at 20 °C to 25 °C (68 ºF to 77 ºF) [see USP Controlled Room Temperature], with excursions permitted to 15 ºC to 30 ºC (59 ºF to 86 ºF) and avoid prolonged exposure to light.
How Supplied
RYANODEX ® (NDC 42367-540-32) is available in 20 mL vials containing a sterile lyophilized mixture of 250 mg dantrolene sodium for reconstitution with 5 mL sterile water for injection USP (without a bacteriostatic agent) to yield an orange colored injectable suspension. Store unreconstituted product at 20 °C to 25 °C (68 ºF to 77 ºF) [see USP Controlled Room Temperature], with excursions permitted to 15 ºC to 30 ºC (59 ºF to 86 ºF) and avoid prolonged exposure to light.
Description
RYANODEX ® is indicated for the: Treatment of malignant hyperthermia in conjunction with appropriate supportive measures [see Dosage and Administration ( 2.1 )] Prevention of malignant hyperthermia in patients at high risk.
Section 42229-5
Pulmonary Edema
There have been reports of pulmonary edema developing during the treatment of malignant hyperthermia crises with another dantrolene sodium dosage form. The contributory effect of the diluent volume and mannitol in these cases is not known.
Thrombophlebitis and Tissue Necrosis
There have been reports of thrombophlebitis following administration of intravenous dantrolene. Tissue necrosis secondary to extravasation has been reported [see Warnings and Precautions (5.3)].
Hypersensitivity/Anaphylactic Reactions
There have been reports of urticaria and erythema possibly associated with the administration of dantrolene sodium for injection. Anaphylaxis has been reported.
Injection Site Reactions
Injection site reactions including pain, erythema, and swelling, commonly due to extravasation, have been reported.
Hepatotoxicity
Cases of hepatotoxicity following the use of intravenous dantrolene products have been reported. Elevated liver enzymes have occurred hours to days following use of intravenous dantrolene, though many of these cases were observed in patients with comorbidities (e.g., critical illness).
8.2 Lactation
Risk Summary
Dantrolene is reported to be present in human milk following intravenous administration over 3 days. There are no data on the effects on the breastfed infant or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed infant, including respiratory depression and muscle weakness, advise patients that breastfeeding is not recommended during treatment with RYANODEX, and for 3 days after the last dose.
Clinical Considerations
A lactating woman should interrupt breastfeeding and pump and discard breast milk during treatment and for 3 days after RYANODEX administration.
11 Description
RYANODEX® (dantrolene sodium) for injectable suspension is a sterile lyophilized powder. RYANODEX is supplied in 20 mL vials containing 250 mg dantrolene sodium and the following inactive ingredients: 125 mg mannitol, 25 mg polysorbate 80, 4 mg povidone K12 and sufficient sodium hydroxide or hydrochloric acid for pH adjustment. When reconstituted with 5 mL sterile water for injection USP (without a bacteriostatic agent), this yields a suspension with a pH of approximately 10.3.
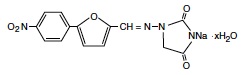
RYANODEX is a skeletal muscle relaxant. Chemically, RYANODEX is a hydrate of 1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2,4-imidazolidinedione sodium salt. The structural formula for the hydrated salt is:
The hydrated salt contains approximately 15% water (3-1/2 moles) and has a molecular weight of 399. The anhydrous salt (dantrolene) has a molecular weight of 336.
8.4 Pediatric Use
The safety and efficacy of RYANODEX in the treatment and prevention of malignant hyperthermia in pediatric patients is based on clinical experience with other intravenous dantrolene sodium products, which suggests adult weight-based doses are appropriate for pediatric patients.
8.5 Geriatric Use
Clinical studies of RYANODEX did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
4 Contraindications
None
5.1 Muscle Weakness
RYANODEX is associated with skeletal muscle weakness. The administration of RYANODEX in human volunteers has been associated with loss of grip strength and weakness in the legs. Patients should not be permitted to ambulate without assistance until they have normal strength and balance.
RYANODEX has been associated with dyspnea, respiratory muscle weakness, and decreased inspiratory capacity. Monitor patients for the adequacy of ventilation.
RYANODEX has been associated with dysphagia. Assess patients for difficulty swallowing and choking.
6 Adverse Reactions
- Administration to conscious subjects is associated with loss of grip strength and weakness in the legs, drowsiness and dizziness.
- Other common adverse reactions are: nausea, thrombophlebitis, tissue necrosis secondary to extravasation, urticaria and erythema, and injection site reactions (pain, erythema, swelling) (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eagle at
1-855-318-2170 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
7 Drug Interactions
7.2 Muscle Relaxants
The concomitant administration of RYANODEX with muscle relaxants may potentiate the neuromuscular block.
12.2 Pharmacodynamics
The administration of intravenous dantrolene to human volunteers was associated with loss of grip strength and weakness in the legs, as well as subjective CNS complaints [see Warnings and Precautions (5.1)].
12.3 Pharmacokinetics
The pharmacokinetics of RYANODEX was investigated in healthy volunteers following single-dose administration as an intravenous push over 60 seconds (dose range of 1 to 2.5 mg/kg), via a peripheral catheter. There was a dose-proportional increase in plasma exposure of dantrolene and its metabolite, 5-hydroxydantrolene. Table 2 presents pharmacokinetic parameters of dantrolene after administration of a single RYANODEX dose of 2.5 mg/kg. Time to peak dantrolene concentration was observed at the first time point collected (i.e., median Tmax is 1 minute post-dose). The mean half-life (t1/2) for dantrolene was independent of the RYANODEX dose administered and ranged from 8.5 hours to 11.4 hours over the 1 to 2.5 mg/kg dose range.
|
1 n=15; single-dose |
|||||
| RYANODEX dose1 |
Cmax
(μg/mL) |
AUC0-inf
(μg*h/mL) |
Half-life (hrs) |
Clearance (L/hr) |
Volume of distribution (L) |
| 2.5 mg/kg | 9.0 ± 4.6 | 77.7 ± 23.2 | 10.8 ± 2.2 | 2.5 ± 1.0 | 36.4 ± 11.7 |
When prophylactic intravenous dantrolene infusion was administered, whole blood dantrolene concentrations remained at a near steady state level for 3 or more hours after the infusion was completed.
1 Indications and Usage
RYANODEX® is indicated for the:
- Treatment of malignant hyperthermia in conjunction with appropriate supportive measures [see Dosage and Administration (2.1)]
- Prevention of malignant hyperthermia in patients at high risk.
10.1 Overdosage Symptoms
Overdosage symptoms include, but are not limited to, muscular weakness and alterations in the state of consciousness (e.g., lethargy, coma), vomiting, diarrhea, and crystalluria.
12.1 Mechanism of Action
In isolated nerve-muscle preparation, dantrolene has been shown to produce relaxation by affecting the contractile response of the muscle at a site beyond the myoneural junction. In skeletal muscle, dantrolene dissociates the excitation-contraction coupling, probably by interfering with the release of Ca++ from the sarcoplasmic reticulum.
In the anesthetic-induced malignant hyperthermia syndrome, evidence points to an intrinsic abnormality of skeletal muscle tissue. In affected humans, it has been postulated that “triggering agents” (e.g., general anesthetics and depolarizing neuromuscular blocking agents) produce a change within the cell which results in an elevated myoplasmic calcium. This elevated myoplasmic calcium activates acute cellular catabolic processes that cascade to the malignant hyperthermia crisis.
The addition of dantrolene to the “triggered” malignant hyperthermic muscle cell may reestablish a normal level of ionized calcium in the myoplasm. Inhibition of calcium release from the sarcoplasmic reticulum by dantrolene reestablishes the myoplasmic calcium equilibrium, increasing the percentage of bound calcium. In this way, physiologic, metabolic, and biochemical changes associated with the malignant hyperthermia crisis may be reversed or attenuated.
5 Warnings and Precautions
- Skeletal Muscle Weakness: Ambulate patients with assistance until they have normal strength and balance (5.1)
- Dyspnea, Respiratory Muscle Weakness, and Decreased Inspiratory Capacity: Monitor patients for the adequacy of ventilation (5.1)
- Dysphagia: Assess patients for difficulty swallowing and choking (5.1)
- Somnolence and Dizziness: Can occur following RYANODEX administration; may persist up to 48 hours post-dose. Ambulate patients with assistance until they have normal strength and balance (5.2)
- Tissue Necrosis with Extravasation: Due to high pH of reconstituted RYANODEX for injectable suspension, care must be taken to prevent extravasation into surrounding tissues (5.3)
2 Dosage and Administration
-
Treatment of Malignant Hyperthermia (MH) (2.1)
- Administer by intravenous push at a minimum of 1 mg/kg. If signs continue, administer additional intravenous boluses up to maximum cumulative dosage of 10 mg/kg.
- Institute supportive measures (e.g., discontinue MH-triggering agents, manage metabolic acidosis, cooling if necessary, administer diuretics)
-
Prevention of MH in Patients at High Risk (2.2)
- Administer 2.5 mg/kg intravenously over a period of at least 1 minute, starting approximately 75 minutes prior to surgery.
- Avoid agents that trigger MH.
- Administer additional individualized doses during anesthesia and surgery if surgery is prolonged.
- Pediatric Patients: recommended weight-based dose is the same as for adults (2.3)
- Reconstitution: With 5 mL of sterile water for injection (without a bacteriostatic agent) prior to administration (2.4)
3 Dosage Forms and Strengths
For injectable suspension: RYANODEX is a sterile, lyophilized powder containing 250 mg of dantrolene sodium for reconstitution, in single-dose vials
5.2 Somnolence and Dizziness
Somnolence and dizziness can occur following administration of RYANODEX and may persist up to 48 hours post-dose. Patients should not be permitted to ambulate without assistance until they have normal strength and balance. Patients must not operate an automobile or engage in other hazardous activities for 48 hours post-dose.
The concomitant use of sedative agents with RYANODEX may increase the risk of somnolence and dizziness.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of another formulation of dantrolene sodium for injection. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
7.1 Calcium Channel Blockers
Cardiovascular collapse in association with marked hyperkalemia has been reported in patients receiving dantrolene in combination with calcium channel blockers. The concomitant use of RYANODEX and calcium channel blockers is not recommended during the treatment of malignant hyperthermia.
10.2 Management of Overdosage
Employ general supportive measures for acute overdosage of RYANODEX.
Administer intravenous fluids in fairly large quantities to avert the possibility of crystalluria. Maintain an adequate airway and keep artificial resuscitation equipment available. Institute electrocardiographic monitoring and carefully observe the patient. The value of dialysis in RYANODEX overdosage is not known.
8 Use in Specific Populations
- Pregnancy: Based on animal data, may cause fetal harm); however, RYANODEX administration for malignant hyperthermia may be lifesaving for the pregnant woman and fetus. Treatment should not be withheld due to pregnancy (8.1)
- Lactation: Breastfeeding is not recommended during treatment with RYANODEX and for 3 days after the final dose. (8.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In a study designed to evaluate the safety and tolerability of RYANODEX, healthy volunteers were randomly assigned to receive treatment with RYANODEX or an active comparator at doses ranging from 1 mg/kg to 2.5 mg/kg.
- The RYANODEX dose was infused over the course of 1 minute for each of the doses evaluated.
- The active comparator was an injectable formulation of dantrolene sodium that differed from RYANODEX in that it contained dantrolene sodium and mannitol at concentrations of 0.33 mg/mL and 50 mg/mL, respectively, when reconstituted according to the product's prescribing information. The active comparator was infused at a rate that administered 20 mg of dantrolene per minute for each of the doses evaluated.
Table 1 displays the most common adverse events in this study. These data are not an adequate basis for comparison of the types or frequencies of adverse event types between RYANODEX and the dantrolene sodium comparator.
Adverse events increased in frequency with increasing doses in the trial, but did not differ in frequency between the two treatment groups. RYANODEX-treated subjects were more likely to report immediate adverse events of flushing, dystonia, and dysphagia than those receiving the active comparator.
In all dose groups, hand grip strength declined after dosing. In general, the decline in hand grip strength was more pronounced and occurred more rapidly in the RYANODEX-treated subjects in the 1.0, 1.75, 2.0 and 2.25 mg/kg treatment groups. In the 2.5 mg/kg treatment group, the decline in hand grip strength both in amount and duration was similar between the two treatment groups.
| Number of subjects (%) | ||
|
RYANODEX
[N=30] n (%) |
Dantrolene Sodium Comparator
[N=31] n (%) |
|
| Flushing | 8 (27) | 1 (3) |
| Somnolence | 5 (17) | 4 (13) |
| Dysphonia | 4 (13) | 1 (3) |
| Dysphagia | 3 (10) | 4 (13) |
| Nausea | 3 (10) | 3 (10) |
| Feeling abnormal | 3 (10) | 3 (10) |
| Headache | 1 (3) | 4 (13) |
| Vomiting | 1 (3) | 2 (6) |
| Vision blurred | 1 (3) | 1 (3) |
| Pain in extremity | 1 (3) | 1 (3) |
| Muscular weakness/Asthenia | 1 (3) | 1 (3) |
| Atrioventricular block | 1 (3) | 0 |
| Tachycardia | 1 (3) | 0 |
| Infusion site pain | 1 (3) | 0 |
| Dizziness | 1 (3) | 0 |
17 Patient Counseling Information
Inform patients, their families, or their caregivers of the following:
Muscle Weakness
Muscle weakness (i.e., decrease in grip strength and weakness of leg muscles, especially walking down stairs) is likely to occur with the use of RYANODEX. Patients should be provided assistance with standing and walking until their strength has returned to normal [see Warnings and Precautions (5.1)].
Difficulty Swallowing
Caution is indicated at meals on the day of administration because difficulty swallowing and choking have occurred with the use of dantrolene sodium products in general; dysphagia has been reported with the use of RYANODEX [see Warnings and Precautions (5.1)].
Dizziness and Somnolence
The use of RYANODEX has been associated with dizziness and somnolence. [see Warnings and Precautions (5.2)].
Driving or Operating Machinery
Symptoms such as “lightheadedness” may occur. Since some of these symptoms may persist for up to 48 hours, patients must not operate an automobile or engage in other hazardous activity during this time [see Warnings and Precautions (5.2)].
Lactation
Advise females not to breastfeed during treatment with RYANODEX and for 3 days after the final dose [see Use in Specific Populations (8.2)].
Marketed by:
Eagle Pharmaceuticals, Inc.
Woodcliff Lake, NJ 07677
2.3 Dosage for Pediatric Patients
16 How Supplied/storage and Handling
RYANODEX® (NDC 42367-540-32) is available in 20 mL vials containing a sterile lyophilized mixture of 250 mg dantrolene sodium for reconstitution with 5 mL sterile water for injection USP (without a bacteriostatic agent) to yield an orange colored injectable suspension.
Store unreconstituted product at 20 °C to 25 °C (68 ºF to 77 ºF) [see USP Controlled Room Temperature], with excursions permitted to 15 ºC to 30 ºC (59 ºF to 86 ºF) and avoid prolonged exposure to light.
7.3 Antipsychotics and Antianxiety Agents
The concomitant administration of RYANODEX with antipsychotic and antianxiety agents may potentiate their effects on the central nervous system [see Warnings and Precautions (5.2)].
2.1 Dosage for Treatment of Malignant Hyperthermia
In addition to RYANODEX treatment, institute the following supportive measures:
- Discontinue use of malignant hyperthermia (MH)-triggering anesthetic agents (i.e., volatile anesthetic gases and succinylcholine).
- Manage the metabolic acidosis
- Institute cooling when necessary
- Administer diuretics to prevent late kidney injury due to myoglobinuria (the amount of mannitol in RYANODEX is insufficient to maintain diuresis) [see Description (11)]
Administer RYANODEX by intravenous push at a minimum dose of 1 mg/kg. If the physiologic and metabolic abnormalities of MH continue, administer additional intravenous boluses up to the maximum cumulative dosage of 10 mg/kg.
If the physiologic and metabolic abnormalities reappear, repeat RYANODEX dosing by intravenous push starting with 1 mg/kg.
2.4 Reconstitution and Administration Instructions
The supplied lyophilized powder must be reconstituted prior to administration:
-
(a)Reconstitute each vial of RYANODEX lyophilized powder by adding 5 mL of sterile water for injection (without a bacteriostatic agent). Do not reconstitute with any other solution (e.g., 5% dextrose injection, 0.9% sodium chloride injection).
-
(b)Shake the vial to ensure an orange-colored uniform suspension. Visually inspect the vial for particulate matter and discoloration prior to administration.
-
(c)Must use the contents of the vial within 6 hours after reconstitution. Store reconstituted suspensions at controlled room temperature (68°F to 77°F or 20°C to 25°C).
Do not dilute or transfer the reconstituted RYANODEX suspension to another container to infuse the product.
Administer the reconstituted RYANODEX suspension either:
- Into the intravenous catheter while an intravenous infusion of 0.9% sodium chloride injection, or 5% dextrose injection is freely running; or
- Into the indwelling catheter - after assuring its patency - without a freely running infusion. Flush the line to assure that there is no residual RYANODEX remaining in the catheter [see Warnings and Precautions (5.3)].
2.2 Dosage for Prevention of Malignant Hyperthermia
The recommended prophylactic dose of RYANODEX is 2.5 mg/kg administered intravenously over a period of at least 1 minute, starting approximately 75 minutes prior to surgery. Avoid agents that trigger MH.
If surgery is prolonged, administer additional individualized RYANODEX doses during anesthesia and surgery [see Clinical Pharmacology (12.3)].
5.3 Potential for Tissue Necrosis With Extravasation
Care must be taken to prevent extravasation of RYANODEX into the surrounding tissues due to the high pH of the reconstituted RYANODEX suspension and potential for tissue necrosis.
Principal Display Panel Ndc: 42367 540 32 Vial Label
NDC 42367-540-32
Sterile
Single Use Only -
Discard Unused Portion
Rx only
Ryanodex®
(dantrolene sodium)
for injectable suspension
250 mg per vial
Reconstitution yields 50 mg/mL
For treatment of malignant hyperthermia, along with the appropriate supportive measures.
For Intravenous Use Only
Reconstitute with Sterile Water for Injection, USP
Principal Display Panel Ndc: 42367 540 32 Carton Label
NDC 42367-540-32
Sterile
Single Use Only -
Discard Unused Portion
Rx only
Ryanodex®
(dantrolene sodium)
for injectable suspension
250 mg per vial
Reconstitution yields 50 mg/mL
For treatment of malignant hyperthermia, along with the appropriate supportive measures.
For intravenous use only
Reconstitute with Sterile Water for Injection, USP
SEE PACKAGE INSERT FOR COMPLETE PREPARATION INSTRUCTIONS AND PRESCRIBING INFORMATION.
Structured Label Content
Section 42229-5 (42229-5)
Pulmonary Edema
There have been reports of pulmonary edema developing during the treatment of malignant hyperthermia crises with another dantrolene sodium dosage form. The contributory effect of the diluent volume and mannitol in these cases is not known.
Thrombophlebitis and Tissue Necrosis
There have been reports of thrombophlebitis following administration of intravenous dantrolene. Tissue necrosis secondary to extravasation has been reported [see Warnings and Precautions (5.3)].
Hypersensitivity/Anaphylactic Reactions
There have been reports of urticaria and erythema possibly associated with the administration of dantrolene sodium for injection. Anaphylaxis has been reported.
Injection Site Reactions
Injection site reactions including pain, erythema, and swelling, commonly due to extravasation, have been reported.
Hepatotoxicity
Cases of hepatotoxicity following the use of intravenous dantrolene products have been reported. Elevated liver enzymes have occurred hours to days following use of intravenous dantrolene, though many of these cases were observed in patients with comorbidities (e.g., critical illness).
8.2 Lactation
Risk Summary
Dantrolene is reported to be present in human milk following intravenous administration over 3 days. There are no data on the effects on the breastfed infant or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed infant, including respiratory depression and muscle weakness, advise patients that breastfeeding is not recommended during treatment with RYANODEX, and for 3 days after the last dose.
Clinical Considerations
A lactating woman should interrupt breastfeeding and pump and discard breast milk during treatment and for 3 days after RYANODEX administration.
11 Description (11 DESCRIPTION)
RYANODEX® (dantrolene sodium) for injectable suspension is a sterile lyophilized powder. RYANODEX is supplied in 20 mL vials containing 250 mg dantrolene sodium and the following inactive ingredients: 125 mg mannitol, 25 mg polysorbate 80, 4 mg povidone K12 and sufficient sodium hydroxide or hydrochloric acid for pH adjustment. When reconstituted with 5 mL sterile water for injection USP (without a bacteriostatic agent), this yields a suspension with a pH of approximately 10.3.
RYANODEX is a skeletal muscle relaxant. Chemically, RYANODEX is a hydrate of 1-[[[5-(4-nitrophenyl)-2-furanyl]methylene]amino]-2,4-imidazolidinedione sodium salt. The structural formula for the hydrated salt is:
The hydrated salt contains approximately 15% water (3-1/2 moles) and has a molecular weight of 399. The anhydrous salt (dantrolene) has a molecular weight of 336.
8.4 Pediatric Use
The safety and efficacy of RYANODEX in the treatment and prevention of malignant hyperthermia in pediatric patients is based on clinical experience with other intravenous dantrolene sodium products, which suggests adult weight-based doses are appropriate for pediatric patients.
8.5 Geriatric Use
Clinical studies of RYANODEX did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
4 Contraindications (4 CONTRAINDICATIONS)
None
5.1 Muscle Weakness
RYANODEX is associated with skeletal muscle weakness. The administration of RYANODEX in human volunteers has been associated with loss of grip strength and weakness in the legs. Patients should not be permitted to ambulate without assistance until they have normal strength and balance.
RYANODEX has been associated with dyspnea, respiratory muscle weakness, and decreased inspiratory capacity. Monitor patients for the adequacy of ventilation.
RYANODEX has been associated with dysphagia. Assess patients for difficulty swallowing and choking.
6 Adverse Reactions (6 ADVERSE REACTIONS)
- Administration to conscious subjects is associated with loss of grip strength and weakness in the legs, drowsiness and dizziness.
- Other common adverse reactions are: nausea, thrombophlebitis, tissue necrosis secondary to extravasation, urticaria and erythema, and injection site reactions (pain, erythema, swelling) (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eagle at
1-855-318-2170 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
7 Drug Interactions (7 DRUG INTERACTIONS)
7.2 Muscle Relaxants
The concomitant administration of RYANODEX with muscle relaxants may potentiate the neuromuscular block.
12.2 Pharmacodynamics
The administration of intravenous dantrolene to human volunteers was associated with loss of grip strength and weakness in the legs, as well as subjective CNS complaints [see Warnings and Precautions (5.1)].
12.3 Pharmacokinetics
The pharmacokinetics of RYANODEX was investigated in healthy volunteers following single-dose administration as an intravenous push over 60 seconds (dose range of 1 to 2.5 mg/kg), via a peripheral catheter. There was a dose-proportional increase in plasma exposure of dantrolene and its metabolite, 5-hydroxydantrolene. Table 2 presents pharmacokinetic parameters of dantrolene after administration of a single RYANODEX dose of 2.5 mg/kg. Time to peak dantrolene concentration was observed at the first time point collected (i.e., median Tmax is 1 minute post-dose). The mean half-life (t1/2) for dantrolene was independent of the RYANODEX dose administered and ranged from 8.5 hours to 11.4 hours over the 1 to 2.5 mg/kg dose range.
|
1 n=15; single-dose |
|||||
| RYANODEX dose1 |
Cmax
(μg/mL) |
AUC0-inf
(μg*h/mL) |
Half-life (hrs) |
Clearance (L/hr) |
Volume of distribution (L) |
| 2.5 mg/kg | 9.0 ± 4.6 | 77.7 ± 23.2 | 10.8 ± 2.2 | 2.5 ± 1.0 | 36.4 ± 11.7 |
When prophylactic intravenous dantrolene infusion was administered, whole blood dantrolene concentrations remained at a near steady state level for 3 or more hours after the infusion was completed.
1 Indications and Usage (1 INDICATIONS AND USAGE)
RYANODEX® is indicated for the:
- Treatment of malignant hyperthermia in conjunction with appropriate supportive measures [see Dosage and Administration (2.1)]
- Prevention of malignant hyperthermia in patients at high risk.
10.1 Overdosage Symptoms
Overdosage symptoms include, but are not limited to, muscular weakness and alterations in the state of consciousness (e.g., lethargy, coma), vomiting, diarrhea, and crystalluria.
12.1 Mechanism of Action
In isolated nerve-muscle preparation, dantrolene has been shown to produce relaxation by affecting the contractile response of the muscle at a site beyond the myoneural junction. In skeletal muscle, dantrolene dissociates the excitation-contraction coupling, probably by interfering with the release of Ca++ from the sarcoplasmic reticulum.
In the anesthetic-induced malignant hyperthermia syndrome, evidence points to an intrinsic abnormality of skeletal muscle tissue. In affected humans, it has been postulated that “triggering agents” (e.g., general anesthetics and depolarizing neuromuscular blocking agents) produce a change within the cell which results in an elevated myoplasmic calcium. This elevated myoplasmic calcium activates acute cellular catabolic processes that cascade to the malignant hyperthermia crisis.
The addition of dantrolene to the “triggered” malignant hyperthermic muscle cell may reestablish a normal level of ionized calcium in the myoplasm. Inhibition of calcium release from the sarcoplasmic reticulum by dantrolene reestablishes the myoplasmic calcium equilibrium, increasing the percentage of bound calcium. In this way, physiologic, metabolic, and biochemical changes associated with the malignant hyperthermia crisis may be reversed or attenuated.
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Skeletal Muscle Weakness: Ambulate patients with assistance until they have normal strength and balance (5.1)
- Dyspnea, Respiratory Muscle Weakness, and Decreased Inspiratory Capacity: Monitor patients for the adequacy of ventilation (5.1)
- Dysphagia: Assess patients for difficulty swallowing and choking (5.1)
- Somnolence and Dizziness: Can occur following RYANODEX administration; may persist up to 48 hours post-dose. Ambulate patients with assistance until they have normal strength and balance (5.2)
- Tissue Necrosis with Extravasation: Due to high pH of reconstituted RYANODEX for injectable suspension, care must be taken to prevent extravasation into surrounding tissues (5.3)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
-
Treatment of Malignant Hyperthermia (MH) (2.1)
- Administer by intravenous push at a minimum of 1 mg/kg. If signs continue, administer additional intravenous boluses up to maximum cumulative dosage of 10 mg/kg.
- Institute supportive measures (e.g., discontinue MH-triggering agents, manage metabolic acidosis, cooling if necessary, administer diuretics)
-
Prevention of MH in Patients at High Risk (2.2)
- Administer 2.5 mg/kg intravenously over a period of at least 1 minute, starting approximately 75 minutes prior to surgery.
- Avoid agents that trigger MH.
- Administer additional individualized doses during anesthesia and surgery if surgery is prolonged.
- Pediatric Patients: recommended weight-based dose is the same as for adults (2.3)
- Reconstitution: With 5 mL of sterile water for injection (without a bacteriostatic agent) prior to administration (2.4)
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
For injectable suspension: RYANODEX is a sterile, lyophilized powder containing 250 mg of dantrolene sodium for reconstitution, in single-dose vials
5.2 Somnolence and Dizziness
Somnolence and dizziness can occur following administration of RYANODEX and may persist up to 48 hours post-dose. Patients should not be permitted to ambulate without assistance until they have normal strength and balance. Patients must not operate an automobile or engage in other hazardous activities for 48 hours post-dose.
The concomitant use of sedative agents with RYANODEX may increase the risk of somnolence and dizziness.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of another formulation of dantrolene sodium for injection. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
7.1 Calcium Channel Blockers
Cardiovascular collapse in association with marked hyperkalemia has been reported in patients receiving dantrolene in combination with calcium channel blockers. The concomitant use of RYANODEX and calcium channel blockers is not recommended during the treatment of malignant hyperthermia.
10.2 Management of Overdosage
Employ general supportive measures for acute overdosage of RYANODEX.
Administer intravenous fluids in fairly large quantities to avert the possibility of crystalluria. Maintain an adequate airway and keep artificial resuscitation equipment available. Institute electrocardiographic monitoring and carefully observe the patient. The value of dialysis in RYANODEX overdosage is not known.
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
- Pregnancy: Based on animal data, may cause fetal harm); however, RYANODEX administration for malignant hyperthermia may be lifesaving for the pregnant woman and fetus. Treatment should not be withheld due to pregnancy (8.1)
- Lactation: Breastfeeding is not recommended during treatment with RYANODEX and for 3 days after the final dose. (8.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In a study designed to evaluate the safety and tolerability of RYANODEX, healthy volunteers were randomly assigned to receive treatment with RYANODEX or an active comparator at doses ranging from 1 mg/kg to 2.5 mg/kg.
- The RYANODEX dose was infused over the course of 1 minute for each of the doses evaluated.
- The active comparator was an injectable formulation of dantrolene sodium that differed from RYANODEX in that it contained dantrolene sodium and mannitol at concentrations of 0.33 mg/mL and 50 mg/mL, respectively, when reconstituted according to the product's prescribing information. The active comparator was infused at a rate that administered 20 mg of dantrolene per minute for each of the doses evaluated.
Table 1 displays the most common adverse events in this study. These data are not an adequate basis for comparison of the types or frequencies of adverse event types between RYANODEX and the dantrolene sodium comparator.
Adverse events increased in frequency with increasing doses in the trial, but did not differ in frequency between the two treatment groups. RYANODEX-treated subjects were more likely to report immediate adverse events of flushing, dystonia, and dysphagia than those receiving the active comparator.
In all dose groups, hand grip strength declined after dosing. In general, the decline in hand grip strength was more pronounced and occurred more rapidly in the RYANODEX-treated subjects in the 1.0, 1.75, 2.0 and 2.25 mg/kg treatment groups. In the 2.5 mg/kg treatment group, the decline in hand grip strength both in amount and duration was similar between the two treatment groups.
| Number of subjects (%) | ||
|
RYANODEX
[N=30] n (%) |
Dantrolene Sodium Comparator
[N=31] n (%) |
|
| Flushing | 8 (27) | 1 (3) |
| Somnolence | 5 (17) | 4 (13) |
| Dysphonia | 4 (13) | 1 (3) |
| Dysphagia | 3 (10) | 4 (13) |
| Nausea | 3 (10) | 3 (10) |
| Feeling abnormal | 3 (10) | 3 (10) |
| Headache | 1 (3) | 4 (13) |
| Vomiting | 1 (3) | 2 (6) |
| Vision blurred | 1 (3) | 1 (3) |
| Pain in extremity | 1 (3) | 1 (3) |
| Muscular weakness/Asthenia | 1 (3) | 1 (3) |
| Atrioventricular block | 1 (3) | 0 |
| Tachycardia | 1 (3) | 0 |
| Infusion site pain | 1 (3) | 0 |
| Dizziness | 1 (3) | 0 |
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Inform patients, their families, or their caregivers of the following:
Muscle Weakness
Muscle weakness (i.e., decrease in grip strength and weakness of leg muscles, especially walking down stairs) is likely to occur with the use of RYANODEX. Patients should be provided assistance with standing and walking until their strength has returned to normal [see Warnings and Precautions (5.1)].
Difficulty Swallowing
Caution is indicated at meals on the day of administration because difficulty swallowing and choking have occurred with the use of dantrolene sodium products in general; dysphagia has been reported with the use of RYANODEX [see Warnings and Precautions (5.1)].
Dizziness and Somnolence
The use of RYANODEX has been associated with dizziness and somnolence. [see Warnings and Precautions (5.2)].
Driving or Operating Machinery
Symptoms such as “lightheadedness” may occur. Since some of these symptoms may persist for up to 48 hours, patients must not operate an automobile or engage in other hazardous activity during this time [see Warnings and Precautions (5.2)].
Lactation
Advise females not to breastfeed during treatment with RYANODEX and for 3 days after the final dose [see Use in Specific Populations (8.2)].
Marketed by:
Eagle Pharmaceuticals, Inc.
Woodcliff Lake, NJ 07677
2.3 Dosage for Pediatric Patients
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
RYANODEX® (NDC 42367-540-32) is available in 20 mL vials containing a sterile lyophilized mixture of 250 mg dantrolene sodium for reconstitution with 5 mL sterile water for injection USP (without a bacteriostatic agent) to yield an orange colored injectable suspension.
Store unreconstituted product at 20 °C to 25 °C (68 ºF to 77 ºF) [see USP Controlled Room Temperature], with excursions permitted to 15 ºC to 30 ºC (59 ºF to 86 ºF) and avoid prolonged exposure to light.
7.3 Antipsychotics and Antianxiety Agents
The concomitant administration of RYANODEX with antipsychotic and antianxiety agents may potentiate their effects on the central nervous system [see Warnings and Precautions (5.2)].
2.1 Dosage for Treatment of Malignant Hyperthermia
In addition to RYANODEX treatment, institute the following supportive measures:
- Discontinue use of malignant hyperthermia (MH)-triggering anesthetic agents (i.e., volatile anesthetic gases and succinylcholine).
- Manage the metabolic acidosis
- Institute cooling when necessary
- Administer diuretics to prevent late kidney injury due to myoglobinuria (the amount of mannitol in RYANODEX is insufficient to maintain diuresis) [see Description (11)]
Administer RYANODEX by intravenous push at a minimum dose of 1 mg/kg. If the physiologic and metabolic abnormalities of MH continue, administer additional intravenous boluses up to the maximum cumulative dosage of 10 mg/kg.
If the physiologic and metabolic abnormalities reappear, repeat RYANODEX dosing by intravenous push starting with 1 mg/kg.
2.4 Reconstitution and Administration Instructions
The supplied lyophilized powder must be reconstituted prior to administration:
-
(a)Reconstitute each vial of RYANODEX lyophilized powder by adding 5 mL of sterile water for injection (without a bacteriostatic agent). Do not reconstitute with any other solution (e.g., 5% dextrose injection, 0.9% sodium chloride injection).
-
(b)Shake the vial to ensure an orange-colored uniform suspension. Visually inspect the vial for particulate matter and discoloration prior to administration.
-
(c)Must use the contents of the vial within 6 hours after reconstitution. Store reconstituted suspensions at controlled room temperature (68°F to 77°F or 20°C to 25°C).
Do not dilute or transfer the reconstituted RYANODEX suspension to another container to infuse the product.
Administer the reconstituted RYANODEX suspension either:
- Into the intravenous catheter while an intravenous infusion of 0.9% sodium chloride injection, or 5% dextrose injection is freely running; or
- Into the indwelling catheter - after assuring its patency - without a freely running infusion. Flush the line to assure that there is no residual RYANODEX remaining in the catheter [see Warnings and Precautions (5.3)].
2.2 Dosage for Prevention of Malignant Hyperthermia
The recommended prophylactic dose of RYANODEX is 2.5 mg/kg administered intravenously over a period of at least 1 minute, starting approximately 75 minutes prior to surgery. Avoid agents that trigger MH.
If surgery is prolonged, administer additional individualized RYANODEX doses during anesthesia and surgery [see Clinical Pharmacology (12.3)].
5.3 Potential for Tissue Necrosis With Extravasation (5.3 Potential for Tissue Necrosis with Extravasation)
Care must be taken to prevent extravasation of RYANODEX into the surrounding tissues due to the high pH of the reconstituted RYANODEX suspension and potential for tissue necrosis.
Principal Display Panel Ndc: 42367 540 32 Vial Label (PRINCIPAL DISPLAY PANEL - NDC: 42367-540-32 - Vial Label)
NDC 42367-540-32
Sterile
Single Use Only -
Discard Unused Portion
Rx only
Ryanodex®
(dantrolene sodium)
for injectable suspension
250 mg per vial
Reconstitution yields 50 mg/mL
For treatment of malignant hyperthermia, along with the appropriate supportive measures.
For Intravenous Use Only
Reconstitute with Sterile Water for Injection, USP
Principal Display Panel Ndc: 42367 540 32 Carton Label (PRINCIPAL DISPLAY PANEL - NDC: 42367-540-32 - Carton Label)
NDC 42367-540-32
Sterile
Single Use Only -
Discard Unused Portion
Rx only
Ryanodex®
(dantrolene sodium)
for injectable suspension
250 mg per vial
Reconstitution yields 50 mg/mL
For treatment of malignant hyperthermia, along with the appropriate supportive measures.
For intravenous use only
Reconstitute with Sterile Water for Injection, USP
SEE PACKAGE INSERT FOR COMPLETE PREPARATION INSTRUCTIONS AND PRESCRIBING INFORMATION.
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:43:34.620939 · Updated: 2026-03-14T22:16:20.175496