cabergoline tablets, usp
89ec147b-a6fe-49bb-9af5-52fc2ce5db7f
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
Cabergoline Tablets, USP are indicated for the treatment of hyperprolactinemic disorders, either idiopathic or due to pituitary adenomas.
Dosage and Administration
The recommended dosage of Cabergoline Tablets, USP for initiation of therapy is 0.25 mg twice a week. Dosage may be increased by 0.25 mg twice weekly up to a dosage of 1 mg twice a week according to the patient's serum prolactin level. Before initiating treatment, cardiovascular evaluation should be performed and echocardiography should be considered to assess for valvular disease. Dosage increases should not occur more rapidly than every 4 weeks, so that the physician can assess the patient's response to each dosage level. If the patient does not respond adequately, and no additional benefit is observed with higher doses, the lowest dose that achieved maximal response should be used and other therapeutic approaches considered. Patients receiving long term treatment with cabergoline should undergo periodic assessment of their cardiac status and echocardiography should be considered. After a normal serum prolactin level has been maintained for 6 months, cabergoline may be discontinued, with periodic monitoring of the serum prolactin level to determine whether or when treatment with cabergoline should be reinstituted. The durability of efficacy beyond 24 months of therapy with cabergoline has not been established.
Contraindications
Cabergoline Tablets, USP are contraindicated in patients with: Uncontrolled hypertension or known hypersensitivity to ergot derivatives. History of cardiac valvular disorders, as suggested by anatomical evidence of valvulopathy of any valve, determined by pre-treatment evaluation including echocardiographic demonstration of valve leaflet thickening, valve restriction, or mixed valve restriction-stenosis. (See WARNINGS ) History of pulmonary, pericardial, or retroperitoneal fibrotic disorders. (See WARNINGS )
Adverse Reactions
The safety of Cabergoline Tablets, USP has been evaluated in more than 900 patients with hyperprolactinemic disorders. Most adverse events were mild or moderate in severity. In a 4-week, double-blind, placebo-controlled study, treatment consisted of placebo or cabergoline at fixed doses of 0.125, 0.5, 0.75, or 1 mg twice weekly. Doses were halved during the first week. Since a possible dose-related effect was observed for nausea only, the four cabergoline treatment groups have been combined. The incidence of the most common adverse events during the placebo-controlled study is presented in the following table. Incidence of Reported Adverse Events During the 4-week, Double-Blind, Placebo-Controlled Trial * Reported at ≥1% for cabergoline Adverse Event * Cabergoline ( n = 168 ) 0 . 125 to 1 mg two times a week Placebo ( n = 20 ) Number ( percent ) Gastrointestinal Nausea 45 (27) 4 (20) Constipation 16 (10) 0 Abdominal pain 9 (5) 1 (5) Dyspepsia 4 (2) 0 Vomiting 4 (2) 0 Central and Peripheral Nervous System Headache 43 (26) 5 (25) Dizziness 25 (15) 1 (5) Paresthesia 2 (1) 0 Vertigo 2 (1) 0 Body As A Whole Asthenia 15 (9) 2 (10) Fatigue 12 (7) 0 Hot flashes 2 (1) 1 (5) Psychiatric Somnolence 9 (5) 1 (5) Depression 5 (3) 1 (5) Nervousness 4 (2) 0 Autonomic Nervous System Postural hypotension 6 (4) 0 Reproductive - Female Breast pain 2 (1) 0 Dysmenorrhea 2 (1) 0 Vision Abnormal vision 2 (1) 0 In the 8-week, double-blind period of the comparative trial with bromocriptine, cabergoline (at a dose of 0.5 mg twice weekly) was discontinued because of an adverse event in 4 of 221 patients (2%) while bromocriptine (at a dose of 2.5 mg two times a day) was discontinued in 14 of 231 patients (6%). The most common reasons for discontinuation from cabergoline were headache, nausea and vomiting (3, 2, and 2 patients, respectively); the most common reasons for discontinuation from bromocriptine were nausea, vomiting, headache, and dizziness or vertigo (10, 3, 3, and 3 patients, respectively). The incidence of the most common adverse events during the double-blind portion of the comparative trial with bromocriptine is presented in the following table. Incidence of Reported Adverse Events During the 8-week, Double-Blind Period of the Comparative Trial with Bromocriptine *Reported at ≥1% for cabergoline Adverse Event * Cabergoline ( n = 221 ) Bromocriptine ( n = 231 ) Number ( percent ) Gastrointestinal Nausea 63 (29) 100 (43) Constipation 15 (7) 21 (9) Abdominal pain 12 (5) 19 (8) Dyspepsia 11 (5) 16 (7) Vomiting 9 (4) 16 (7) Dry mouth 5 (2) 2 (1) Diarrhea 4 (2) 7 (3) Flatulence 4 (2) 3 (1) Throat irritation 2 (1) 0 Toothache 2 (1) 0 Central and Peripheral Nervous System Headache 58 (26) 62 (27) Dizziness 38 (17) 42 (18) Vertigo 9 (4) 10 (4) Paresthesia 5 (2) 6 (3) Body As A Whole Asthenia 13 (6) 15 (6) Fatigue 10 (5) 18 (8) Syncope 3 (1) 3 (1) Influenza-like symptoms 2 (1) 0 Malaise 2 (1) 0 Periorbital edema 2 (1) 2 (1) Peripheral edema 2 (1) 1 Psychiatric Depression 7 (3) 5 (2) Somnolence 5 (2) 5 (2) Anorexia 3 (1) 3 (1) Anxiety 3 (1) 3 (1) Insomnia 3 (1) 2 (1) Impaired concentration 2 (1) 1 Nervousness 2 (1) 5 (2) Cardiovascular Hot flashes 6 (3) 3 (1) Hypotension 3 (1) 4 (2) Dependent edema 2 (1) 1 Palpitation 2 (1) 5 (2) Reproductive – Female Breast pain 5 (2) 8 (3) Dysmenorrhea 2 (1) 1 Skin and Appendages Acne 3 (1) 0 Pruritus 2 (1) 1 Musculoskeletal Pain 4 (2) 6 (3) Arthralgia 2 (1) 0 Respiratory Rhinitis 2 (1) 9 (4) Vision Abnormal Vision 2 (1) 2 (1) Other adverse events that were reported at an incidence of <1.0% in the overall clinical studies follow. Body as a Whole: facial edema, influenza-like symptoms, malaise Cardiovascular System: hypotension, syncope, palpitations Digestive System: dry mouth, flatulence, diarrhea, anorexia Metabolic and Nutritional System: weight loss, weight gain Nervous System: somnolence, nervousness, paresthesia, insomnia, anxiety Respiratory System: nasal stuffiness, epistaxis Skin and Appendages: acne, pruritus Special Senses: abnormal vision Urogenital System: dysmenorrhea, increased libido The safety of cabergoline has been evaluated in approximately 1,200 patients with Parkinson's disease in controlled and uncontrolled studies at dosages of up to 11.5 mg/day which greatly exceeds the maximum recommended dosage of cabergoline for hyperprolactinemic disorders. In addition to the adverse events that occurred in the patients with hyperprolactinemic disorders, the most common adverse events in patients with Parkinson's disease were dyskinesia, hallucinations, confusion, and peripheral edema. Heart failure, pleural effusion, pulmonary fibrosis, and gastric or duodenal ulcer occurred rarely. One case of constrictive pericarditis has been reported.
Drug Interactions
Cabergoline should not be administered concurrently with D 2 -antagonists, such as phenothiazines, butyrophenones, thioxanthenes, or metoclopramide.
How Supplied
Product: 50090-5834 NDC: 50090-5834-0 8 TABLET in a BOTTLE
Description
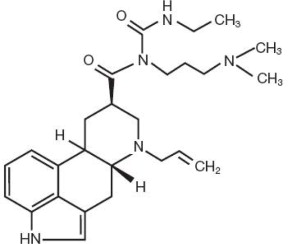
Cabergoline Tablets, USP contain Cabergoline USP a dopamine receptor agonist. The chemical name for Cabergoline USP is 1-[(6-allylergolin-8β-yl)-carbonyl]-1-[3-(dimethylamino)propyl]-3-ethylurea. Its molecular formula is C 26 H 37 N 5 O 2 , and its molecular weight is 451.62. The structural formula is as follows: Cabergoline USP is a white powder soluble in ethyl alcohol, chloroform, and N, N-dimethylformamide (DMF); slightly soluble in 0.1N hydrochloric acid; very slightly soluble in n-hexane; and insoluble in water. Cabergoline Tablets, USP for oral administration, contains 0.5 mg of Cabergoline USP. Inactive ingredients consist of microcrystalline cellulose, croscarmellose sodium, citric acid, and magnesium stearate.
Medication Information
Warnings
- Pregnancy: Dopamine agonists in general should not be used in patients with pregnancy-induced hypertension, for example, preeclampsia eclampsia, and post partum hypertension, unless the potential benefit is judged to outweigh the possible risk.
- Fibrotic Complications:
a. Cardiac Valvulopathy:
All patients should undergo a cardiovascular evaluation, including echocardiogram to assess the potential presence of valvular disease. If valvular disease is detected, the patient should not be treated with cabergoline (See Contraindications ). Post marketing cases of cardiac valvulopathy have been reported in patients receiving cabergoline. These cases have generally occurred during administration of high doses of cabergoline (>2 mg/day) for the treatment of Parkinson's disease. Cases of cardiac valvulopathy have also been reported in patients receiving lower doses of cabergoline for the treatment of hyperprolactinemic disorders.
A multi-country, retrospective cohort study using general practice records and record linkage systems in the UK, Italy and the Netherlands was conducted to assess the association between new use of dopamine agonists including cabergoline (n=27,812) for Parkinson's disease and hyperprolactinemia and cardiac valvular regurgitation (CVR), other fibroses, and other cardiopulmonary events over a maximum of 12 years of follow up. In this study, the use of cabergoline among persons with Parkinson's disease was associated with an increased risk of CVR when compared to non-ergot-derived dopamine agonists (DAs) and levodopa [Incidence Rate (IR) per 10,000 person years of 68.1 (95% confidence interval (CI): 37.2 - 115.3) for cabergoline vs. 10.0 (95% CI: 5.2 - 19.4) for non-ergot DAs and 11.3 (95% CI: 7.2 - 17.0) for levodopa]. In the study analysis confined to persons with dopamine agonist-treated hyperprolactinemia (n=8,386), when compared to non-use (n=15,147), persons exposed to cabergoline did not have an elevated risk of CVR. The findings with respect to the risk of CVR associated with cabergoline treatment for persons with Parkinson's disease (increased risk) and those with hyperprolactinemia (no increased risk) are consistent with the findings in other published studies.
Physicians should use the lowest effective dose of cabergoline for the treatment of hyperprolactinemic disorders and should periodically reassess the need for continuing therapy with cabergoline. Following treatment initiation, clinical and diagnostic monitoring (for example, chest x-ray, CT scan and cardiac echocardiogram) should be conducted to assess the risk of cardiac valvulopathy. The recommended frequency of routine echocardiographic monitoring is every 6 to 12 months or as clinically indicated with the presence of signs and symptoms such as edema, new cardiac murmer, dyspnea or congestive heart failure.
Cabergoline should be discontinued if an echocardiogram reveals new valvular regurgitation, valvular restriction or valve leaflet thickening.
Cabergoline should be used with caution in patients exposed to other medications associated with valvulopathy.
b. Extracardiac Fibrotic Reactions:
Postmarketing cases of pleural, pericardial and retroperitoneal fibrosis have been following administration of cabergoline. Some reports were in patients previously treated with other ergotinic dopamine agonists. Cabergoline should not be used in patients with a history of cardiac or extracardiac fibrotic disorders.
Fibrotic disorders can have an insidious onset and patients should be monitored for manifestations of progressive fibrosis. Therefore, during treatment, attention should be paid to the signs and symptoms of:
- Pleuro-pulmonary disease such as dyspnea, shortness of breath, persistent cough or chest pain
- Renal insufficiency or ureteral/abdominal vascular obstruction that may occur with pain in the loin/flank and lower limb edema as well as any possible abdominal masses or tenderness that may indicate retroperitoneal fibrosis.
- Cardiac failure: Cases of valvular and pericardial fibrosis have often manifested as cardiac failure. Therefore, valvular fibrosis (and constrictive pericarditis) should be excluded if such symptoms occur.
Clinical and diagnostic monitoring such as erythrocyte sedimentation rate, chest x-ray, serum creatinine measurements, and other investigations should be considered at baseline and as necessary while patients are treated with cabergoline.
Following diagnosis of pleural effusion or pulmonary fibrosis, the discontinuance of cabergoline was reported to result in improvement of signs and symptoms.
Indications and Usage
Cabergoline Tablets, USP are indicated for the treatment of hyperprolactinemic disorders, either idiopathic or due to pituitary adenomas.
Dosage and Administration
The recommended dosage of Cabergoline Tablets, USP for initiation of therapy is 0.25 mg twice a week. Dosage may be increased by 0.25 mg twice weekly up to a dosage of 1 mg twice a week according to the patient's serum prolactin level. Before initiating treatment, cardiovascular evaluation should be performed and echocardiography should be considered to assess for valvular disease.
Dosage increases should not occur more rapidly than every 4 weeks, so that the physician can assess the patient's response to each dosage level. If the patient does not respond adequately, and no additional benefit is observed with higher doses, the lowest dose that achieved maximal response should be used and other therapeutic approaches considered. Patients receiving long term treatment with cabergoline should undergo periodic assessment of their cardiac status and echocardiography should be considered.
After a normal serum prolactin level has been maintained for 6 months, cabergoline may be discontinued, with periodic monitoring of the serum prolactin level to determine whether or when treatment with cabergoline should be reinstituted. The durability of efficacy beyond 24 months of therapy with cabergoline has not been established.
Contraindications
Cabergoline Tablets, USP are contraindicated in patients with:
- Uncontrolled hypertension or known hypersensitivity to ergot derivatives.
- History of cardiac valvular disorders, as suggested by anatomical evidence of valvulopathy of any valve, determined by pre-treatment evaluation including echocardiographic demonstration of valve leaflet thickening, valve restriction, or mixed valve restriction-stenosis. (See WARNINGS )
- History of pulmonary, pericardial, or retroperitoneal fibrotic disorders. (See WARNINGS )
Adverse Reactions
The safety of Cabergoline Tablets, USP has been evaluated in more than 900 patients with hyperprolactinemic disorders. Most adverse events were mild or moderate in severity.
In a 4-week, double-blind, placebo-controlled study, treatment consisted of placebo or cabergoline at fixed doses of 0.125, 0.5, 0.75, or 1 mg twice weekly. Doses were halved during the first week. Since a possible dose-related effect was observed for nausea only, the four cabergoline treatment groups have been combined. The incidence of the most common adverse events during the placebo-controlled study is presented in the following table.
|
*Reported at ≥1% for cabergoline |
||
|
Adverse
Event
*
|
Cabergoline
( n = 168 ) 0 . 125 to 1 mg two times a week |
Placebo
(
n
=
20
)
|
|
|
Number
(
percent
)
|
|
|
Gastrointestinal
|
|
|
| Nausea |
45 (27) |
4 (20) |
| Constipation |
16 (10) |
0 |
| Abdominal pain |
9 (5) |
1 (5) |
| Dyspepsia |
4 (2) |
0 |
| Vomiting |
4 (2) |
0 |
|
Central
and
Peripheral
Nervous
System
|
|
|
| Headache |
43 (26) |
5 (25) |
| Dizziness |
25 (15) |
1 (5) |
| Paresthesia |
2 (1) |
0 |
| Vertigo |
2 (1) |
0 |
|
Body
As
A
Whole
|
|
|
| Asthenia |
15 (9) |
2 (10) |
| Fatigue |
12 (7) |
0 |
| Hot flashes |
2 (1) |
1 (5) |
|
Psychiatric
|
|
|
| Somnolence |
9 (5) |
1 (5) |
| Depression |
5 (3) |
1 (5) |
| Nervousness |
4 (2) |
0 |
|
Autonomic
Nervous
System
|
|
|
| Postural hypotension |
6 (4) |
0 |
|
Reproductive
-
Female
|
|
|
| Breast pain |
2 (1) |
0 |
| Dysmenorrhea |
2 (1) |
0 |
|
Vision
|
|
|
| Abnormal vision |
2 (1) |
0 |
In the 8-week, double-blind period of the comparative trial with bromocriptine, cabergoline (at a dose of 0.5 mg twice weekly) was discontinued because of an adverse event in 4 of 221 patients (2%) while bromocriptine (at a dose of 2.5 mg two times a day) was discontinued in 14 of 231 patients (6%). The most common reasons for discontinuation from cabergoline were headache, nausea and vomiting (3, 2, and 2 patients, respectively); the most common reasons for discontinuation from bromocriptine were nausea, vomiting, headache, and dizziness or vertigo (10, 3, 3, and 3 patients, respectively). The incidence of the most common adverse events during the double-blind portion of the comparative trial with bromocriptine is presented in the following table.
|
*Reported at ≥1% for cabergoline |
||
|
Adverse
Event
*
|
Cabergoline
( n = 221 ) |
Bromocriptine
( n = 231 ) |
|
|
Number
(
percent
)
|
|
|
Gastrointestinal
|
|
|
| Nausea |
63 (29) |
100 (43) |
| Constipation |
15 (7) |
21 (9) |
| Abdominal pain |
12 (5) |
19 (8) |
| Dyspepsia |
11 (5) |
16 (7) |
| Vomiting |
9 (4) |
16 (7) |
| Dry mouth |
5 (2) |
2 (1) |
| Diarrhea |
4 (2) |
7 (3) |
| Flatulence |
4 (2) |
3 (1) |
| Throat irritation |
2 (1) |
0 |
| Toothache |
2 (1) |
0 |
|
Central
and
Peripheral
Nervous
System
|
|
|
| Headache |
58 (26) |
62 (27) |
| Dizziness |
38 (17) |
42 (18) |
| Vertigo |
9 (4) |
10 (4) |
| Paresthesia |
5 (2) |
6 (3) |
|
Body
As
A
Whole
|
|
|
| Asthenia |
13 (6) |
15 (6) |
| Fatigue |
10 (5) |
18 (8) |
| Syncope |
3 (1) |
3 (1) |
| Influenza-like symptoms |
2 (1) |
0 |
| Malaise |
2 (1) |
0 |
| Periorbital edema |
2 (1) |
2 (1) |
| Peripheral edema |
2 (1) |
1 |
|
Psychiatric
|
|
|
| Depression |
7 (3) |
5 (2) |
| Somnolence |
5 (2) |
5 (2) |
| Anorexia |
3 (1) |
3 (1) |
| Anxiety |
3 (1) |
3 (1) |
| Insomnia |
3 (1) |
2 (1) |
| Impaired concentration |
2 (1) |
1 |
| Nervousness |
2 (1) |
5 (2) |
|
Cardiovascular
|
|
|
| Hot flashes |
6 (3) |
3 (1) |
| Hypotension |
3 (1) |
4 (2) |
| Dependent edema |
2 (1) |
1 |
| Palpitation |
2 (1) |
5 (2) |
|
Reproductive
–
Female
|
|
|
| Breast pain |
5 (2) |
8 (3) |
| Dysmenorrhea |
2 (1) |
1 |
|
Skin
and
Appendages
|
|
|
| Acne |
3 (1) |
0 |
| Pruritus |
2 (1) |
1 |
|
Musculoskeletal
|
|
|
| Pain |
4 (2) |
6 (3) |
| Arthralgia |
2 (1) |
0 |
|
Respiratory
|
|
|
| Rhinitis |
2 (1) |
9 (4) |
|
Vision
|
|
|
| Abnormal Vision |
2 (1) |
2 (1) |
Other adverse events that were reported at an incidence of <1.0% in the overall clinical studies follow.
Body as a Whole: facial edema, influenza-like symptoms, malaise
Cardiovascular System: hypotension, syncope, palpitations
Digestive System: dry mouth, flatulence, diarrhea, anorexia
Metabolic and Nutritional System: weight loss, weight gain
Nervous System: somnolence, nervousness, paresthesia, insomnia, anxiety
Respiratory System: nasal stuffiness, epistaxis
Skin and Appendages: acne, pruritus
Special Senses: abnormal vision
Urogenital System: dysmenorrhea, increased libido
The safety of cabergoline has been evaluated in approximately 1,200 patients with Parkinson's disease in controlled and uncontrolled studies at dosages of up to 11.5 mg/day which greatly exceeds the maximum recommended dosage of cabergoline for hyperprolactinemic disorders. In addition to the adverse events that occurred in the patients with hyperprolactinemic disorders, the most common adverse events in patients with Parkinson's disease were dyskinesia, hallucinations, confusion, and peripheral edema. Heart failure, pleural effusion, pulmonary fibrosis, and gastric or duodenal ulcer occurred rarely. One case of constrictive pericarditis has been reported.
Drug Interactions
Cabergoline should not be administered concurrently with D2-antagonists, such as phenothiazines, butyrophenones, thioxanthenes, or metoclopramide.
How Supplied
Product: 50090-5834
NDC: 50090-5834-0 8 TABLET in a BOTTLE
Description
Cabergoline Tablets, USP contain Cabergoline USP a dopamine receptor agonist. The chemical name for Cabergoline USP is 1-[(6-allylergolin-8β-yl)-carbonyl]-1-[3-(dimethylamino)propyl]-3-ethylurea. Its molecular formula is C26H37N5O2, and its molecular weight is 451.62. The structural formula is as follows:
Cabergoline USP is a white powder soluble in ethyl alcohol, chloroform, and N, N-dimethylformamide (DMF); slightly soluble in 0.1N hydrochloric acid; very slightly soluble in n-hexane; and insoluble in water.
Cabergoline Tablets, USP for oral administration, contains 0.5 mg of Cabergoline USP. Inactive ingredients consist of microcrystalline cellulose, croscarmellose sodium, citric acid, and magnesium stearate.
Section 42229-5
Clinical Studies: The prolactin-lowering efficacy of cabergoline was demonstrated in hyperprolactinemic women in two randomized, double-blind, comparative studies, one with placebo and the other with bromocriptine. In the placebo-controlled study (placebo n=20; cabergoline n=168), cabergoline produced a dose-related decrease in serum prolactin levels with prolactin normalized after 4 weeks of treatment in 29%, 76%, 74% and 95% of the patients receiving 0.125, 0.5, 0.75, and 1 mg twice weekly, respectively.
In the 8-week, double-blind period of the comparative trial with bromocriptine (cabergoline n=223; bromocriptine n=236 in the intent-to-treat analysis), prolactin was normalized in 77% of the patients treated with cabergoline at 0.5 mg twice weekly compared with 59% of those treated with bromocriptine at 2.5 mg twice daily. Restoration of menses occurred in 77% of the women treated with cabergoline, compared with 70% of those treated with bromocriptine. Among patients with galactorrhea, this symptom disappeared in 73% of those treated with cabergoline compared with 56% of those treated with bromocriptine.
Section 43679-0
Mechanism of Action: The secretion of prolactin by the anterior pituitary is mainly under hypothalmic inhibitory control, likely exerted through release of dopamine by tuberoinfundibular neurons. Cabergoline is a long-acting dopamine receptor agonist with a high affinity for D2 receptors. Results of in vitro studies demonstrate that cabergoline exerts a direct inhibitory effect on the secretion of prolactin by rat pituitary lactotrophs. Cabergoline decreased serum prolactin levels in reserpinized rats. Receptor-binding studies indicate that cabergoline has low affinity for dopamine D1, α1- and α2-adrenergic, and 5-HT1- and 5-HT2-serotonin receptors.
Overdosage
Overdosage might be expected to produce nasal congestion, syncope, or hallucinations. Measures to support blood pressure should be taken if necessary.
Cabergoline
Geriatric Use
Clinical studies of cabergoline did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Pediatric Use
Safety and effectiveness of cabergoline in pediatric patients have not been established.
Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from cabergoline, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. Use of cabergoline for the inhibition or suppression of physiologic lactation is not recommended (see PRECAUTIONS section).
The prolactin-lowering action of cabergoline suggests that it will interfere with lactation. Due to this interference with lactation, cabergoline should not be given to women postpartum who are breastfeeding or who are planning to breastfeed.
Pharmacodynamics
Dose response with inhibition of plasma prolactin, onset of maximal effect, and duration of effect has been documented following single cabergoline doses to healthy volunteers (0.05 to 1.5 mg) and hyperprolactinemic patients (0.3 to 1 mg). In volunteers, prolactin inhibition was evident at doses >0.2 mg, while doses ≥0.5 mg caused maximal suppression in most subjects. Higher doses produce prolactin suppression in a greater proportion of subjects and with an earlier onset and longer duration of action. In 12 healthy volunteers, 0.5, 1 and 1.5 mg doses resulted in complete prolactin inhibition, with a maximum effect within 3 hours in 92% to 100% of subjects after the 1 and 1.5 mg doses compared with 50% of subjects after the 0.5 mg dose.
In hyperprolactinemic patients (n=51), the maximal prolactin decrease after a 0.6 mg single dose of cabergoline was comparable to 2.5 mg bromocriptine; however, the duration of effect was markedly longer (14 days vs. 24 hours). The time to maximal effect was shorter for bromocriptine than cabergoline (6 hours vs. 48 hours).
In 72 healthy volunteers, single or multiple doses (up to 2 mg) of cabergoline resulted in selective inhibition of prolactin with no apparent effect on other anterior pituitary hormones (GH, FSH, LH, ACTH, and TSH) or cortisol.
General Precautions
General: Initial doses higher than 1 mg may produce orthostatic hypotension. Care should be exercised when administering cabergoline with other medications known to lower blood pressure.
Food Drug Interaction
In 12 healthy adult volunteers, food did not alter cabergoline kinetics.
Information for Patients
Patients should be altered to the possibility that patients may experience intense urges to spend money uncontrollably, intense urges to gamble, increased sexual urges, and other intense urges and the inability to control these urges while taking cabergoline. Advise patients to inform their healthcare provider if they develop new or increased uncontrolled spending, gambling urges, sexual urges, or other urges while being treated with cabergoline [See PRECAUTIONS].
Patients should be instructed to notify their physician if they suspect they are pregnant, become pregnant, or intend to become pregnant during therapy. A pregnancy test should be done if there is any suspicion of pregnancy and continuation of treatment should be discussed with their physician.
Patients should notify their physician if they develop shortness of breath, persistent cough, difficulty with breathing when lying down, or swelling in their extremities.
Pregnancy: Teratogenic Effects: Category B
Reproduction studies have been performed with cabergoline in mice, rats, and rabbits administered by gavage.
(Multiples of the maximum recommended human dose in this section are calculated on a body surface area basis using total mg/m2/week for animals and mg/m2/week for a 50 kg human.)
There were maternotoxic effects but no teratogenic effects in mice given cabergoline at doses up to 8 mg/kg/day (approximately 55 times the maximum recommended human dose) during the period of organogenesis.
A dose of 0.012 mg/kg/day (approximately 1/7 the maximum recommended human dose) during the period of organogenesis in rats caused an increase in post-implantation embryofetal losses. These losses could be due to the prolactin inhibitory properties of cabergoline in rats. At daily doses of 0.5 mg/kg/day (approximately 19 times the maximum recommended human dose) during the period of organogenesis in the rabbit, cabergoline caused maternotoxicity characterized by a loss of body weight and decreased food consumption. Doses of 4 mg/kg/day (approximately 150 times the maximum recommended human dose) during the period of organogenesis in the rabbit caused an increased occurrence of various malformations. However, in another study in rabbits, no treatment-related malformations or embryofetotoxicity were observed at doses up to 8 mg/kg/day (approximately 300 times the maximum recommended human dose).
In rats, doses higher than 0.003 mg/kg/day (approximately 1/28 the maximum recommended human dose) from 6 days before parturition and throughout the lactation period inhibited growth and caused death of offspring due to decreased milk secretion.
There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Carcinogenesis and Mutagenesis and Impairment of Fertility
Carcinogenicity studies were conducted in mice and rats with cabergoline given by gavage at doses up to 0.98 mg/kg/day and 0.32 mg/kg/day, respectively. These doses are 7 times and 4 times the maximum recommended human dose calculated on a body surface area basis using total mg/m2/week in rodents and mg/m2/week for a 50 kg human.
There was a slight increase in the incidence of cervical and uterine leiomyomas and uterine leiomyosarcomas in mice. In rats, there was a slight increase in malignant tumors of the cervix and uterus and interstitial cell adenomas. The occurrence of tumors in female rodents may be related to the prolonged suppression of prolactin secretion because prolactin is needed in rodents for the maintenance of the corpus luteum. In the absence of prolactin, the estrogen/progesterone ratio is increased, thereby increasing the risk for uterine tumors. In male rodents, the decrease in serum prolactin levels was associated with an increase in serum luteinizing hormone, which is thought to be a compensatory effect to maintain testicular steroid synthesis. Since these hormonal mechanisms are thought to be species-specific, the relevance of these tumors to humans is not known.
The mutagenic potential of cabergoline was evaluated and found to be negative in a battery of in vitro tests. These tests included the bacterial mutation (Ames) test with Salmonella typhimurium, the gene mutation assay with Schizosaccharomyces pombe P1 and V79 Chinese hamster cells, DNA damage and repair in Saccharomyces cerevisiae D4 , and chromosomal aberrations in human lymphocytes. Cabergoline was also negative in the bone marrow micronucleus test in the mouse.
In female rats, a daily dose of 0.003 mg/kg for 2 weeks prior to mating and throughout the mating period inhibited conception. This dose represents approximately 1/28 the maximum recommended human dose calculated on a body surface area basis using total mg/m2/week in rats and mg/m2/week for a 50 kg human.
Structured Label Content
Warnings (WARNINGS)
- Pregnancy: Dopamine agonists in general should not be used in patients with pregnancy-induced hypertension, for example, preeclampsia eclampsia, and post partum hypertension, unless the potential benefit is judged to outweigh the possible risk.
- Fibrotic Complications:
a. Cardiac Valvulopathy:
All patients should undergo a cardiovascular evaluation, including echocardiogram to assess the potential presence of valvular disease. If valvular disease is detected, the patient should not be treated with cabergoline (See Contraindications ). Post marketing cases of cardiac valvulopathy have been reported in patients receiving cabergoline. These cases have generally occurred during administration of high doses of cabergoline (>2 mg/day) for the treatment of Parkinson's disease. Cases of cardiac valvulopathy have also been reported in patients receiving lower doses of cabergoline for the treatment of hyperprolactinemic disorders.
A multi-country, retrospective cohort study using general practice records and record linkage systems in the UK, Italy and the Netherlands was conducted to assess the association between new use of dopamine agonists including cabergoline (n=27,812) for Parkinson's disease and hyperprolactinemia and cardiac valvular regurgitation (CVR), other fibroses, and other cardiopulmonary events over a maximum of 12 years of follow up. In this study, the use of cabergoline among persons with Parkinson's disease was associated with an increased risk of CVR when compared to non-ergot-derived dopamine agonists (DAs) and levodopa [Incidence Rate (IR) per 10,000 person years of 68.1 (95% confidence interval (CI): 37.2 - 115.3) for cabergoline vs. 10.0 (95% CI: 5.2 - 19.4) for non-ergot DAs and 11.3 (95% CI: 7.2 - 17.0) for levodopa]. In the study analysis confined to persons with dopamine agonist-treated hyperprolactinemia (n=8,386), when compared to non-use (n=15,147), persons exposed to cabergoline did not have an elevated risk of CVR. The findings with respect to the risk of CVR associated with cabergoline treatment for persons with Parkinson's disease (increased risk) and those with hyperprolactinemia (no increased risk) are consistent with the findings in other published studies.
Physicians should use the lowest effective dose of cabergoline for the treatment of hyperprolactinemic disorders and should periodically reassess the need for continuing therapy with cabergoline. Following treatment initiation, clinical and diagnostic monitoring (for example, chest x-ray, CT scan and cardiac echocardiogram) should be conducted to assess the risk of cardiac valvulopathy. The recommended frequency of routine echocardiographic monitoring is every 6 to 12 months or as clinically indicated with the presence of signs and symptoms such as edema, new cardiac murmer, dyspnea or congestive heart failure.
Cabergoline should be discontinued if an echocardiogram reveals new valvular regurgitation, valvular restriction or valve leaflet thickening.
Cabergoline should be used with caution in patients exposed to other medications associated with valvulopathy.
b. Extracardiac Fibrotic Reactions:
Postmarketing cases of pleural, pericardial and retroperitoneal fibrosis have been following administration of cabergoline. Some reports were in patients previously treated with other ergotinic dopamine agonists. Cabergoline should not be used in patients with a history of cardiac or extracardiac fibrotic disorders.
Fibrotic disorders can have an insidious onset and patients should be monitored for manifestations of progressive fibrosis. Therefore, during treatment, attention should be paid to the signs and symptoms of:
- Pleuro-pulmonary disease such as dyspnea, shortness of breath, persistent cough or chest pain
- Renal insufficiency or ureteral/abdominal vascular obstruction that may occur with pain in the loin/flank and lower limb edema as well as any possible abdominal masses or tenderness that may indicate retroperitoneal fibrosis.
- Cardiac failure: Cases of valvular and pericardial fibrosis have often manifested as cardiac failure. Therefore, valvular fibrosis (and constrictive pericarditis) should be excluded if such symptoms occur.
Clinical and diagnostic monitoring such as erythrocyte sedimentation rate, chest x-ray, serum creatinine measurements, and other investigations should be considered at baseline and as necessary while patients are treated with cabergoline.
Following diagnosis of pleural effusion or pulmonary fibrosis, the discontinuance of cabergoline was reported to result in improvement of signs and symptoms.
Indications and Usage (INDICATIONS AND USAGE)
Cabergoline Tablets, USP are indicated for the treatment of hyperprolactinemic disorders, either idiopathic or due to pituitary adenomas.
Dosage and Administration (DOSAGE AND ADMINISTRATION)
The recommended dosage of Cabergoline Tablets, USP for initiation of therapy is 0.25 mg twice a week. Dosage may be increased by 0.25 mg twice weekly up to a dosage of 1 mg twice a week according to the patient's serum prolactin level. Before initiating treatment, cardiovascular evaluation should be performed and echocardiography should be considered to assess for valvular disease.
Dosage increases should not occur more rapidly than every 4 weeks, so that the physician can assess the patient's response to each dosage level. If the patient does not respond adequately, and no additional benefit is observed with higher doses, the lowest dose that achieved maximal response should be used and other therapeutic approaches considered. Patients receiving long term treatment with cabergoline should undergo periodic assessment of their cardiac status and echocardiography should be considered.
After a normal serum prolactin level has been maintained for 6 months, cabergoline may be discontinued, with periodic monitoring of the serum prolactin level to determine whether or when treatment with cabergoline should be reinstituted. The durability of efficacy beyond 24 months of therapy with cabergoline has not been established.
Contraindications (CONTRAINDICATIONS)
Cabergoline Tablets, USP are contraindicated in patients with:
- Uncontrolled hypertension or known hypersensitivity to ergot derivatives.
- History of cardiac valvular disorders, as suggested by anatomical evidence of valvulopathy of any valve, determined by pre-treatment evaluation including echocardiographic demonstration of valve leaflet thickening, valve restriction, or mixed valve restriction-stenosis. (See WARNINGS )
- History of pulmonary, pericardial, or retroperitoneal fibrotic disorders. (See WARNINGS )
Adverse Reactions (ADVERSE REACTIONS)
The safety of Cabergoline Tablets, USP has been evaluated in more than 900 patients with hyperprolactinemic disorders. Most adverse events were mild or moderate in severity.
In a 4-week, double-blind, placebo-controlled study, treatment consisted of placebo or cabergoline at fixed doses of 0.125, 0.5, 0.75, or 1 mg twice weekly. Doses were halved during the first week. Since a possible dose-related effect was observed for nausea only, the four cabergoline treatment groups have been combined. The incidence of the most common adverse events during the placebo-controlled study is presented in the following table.
|
*Reported at ≥1% for cabergoline |
||
|
Adverse
Event
*
|
Cabergoline
( n = 168 ) 0 . 125 to 1 mg two times a week |
Placebo
(
n
=
20
)
|
|
|
Number
(
percent
)
|
|
|
Gastrointestinal
|
|
|
| Nausea |
45 (27) |
4 (20) |
| Constipation |
16 (10) |
0 |
| Abdominal pain |
9 (5) |
1 (5) |
| Dyspepsia |
4 (2) |
0 |
| Vomiting |
4 (2) |
0 |
|
Central
and
Peripheral
Nervous
System
|
|
|
| Headache |
43 (26) |
5 (25) |
| Dizziness |
25 (15) |
1 (5) |
| Paresthesia |
2 (1) |
0 |
| Vertigo |
2 (1) |
0 |
|
Body
As
A
Whole
|
|
|
| Asthenia |
15 (9) |
2 (10) |
| Fatigue |
12 (7) |
0 |
| Hot flashes |
2 (1) |
1 (5) |
|
Psychiatric
|
|
|
| Somnolence |
9 (5) |
1 (5) |
| Depression |
5 (3) |
1 (5) |
| Nervousness |
4 (2) |
0 |
|
Autonomic
Nervous
System
|
|
|
| Postural hypotension |
6 (4) |
0 |
|
Reproductive
-
Female
|
|
|
| Breast pain |
2 (1) |
0 |
| Dysmenorrhea |
2 (1) |
0 |
|
Vision
|
|
|
| Abnormal vision |
2 (1) |
0 |
In the 8-week, double-blind period of the comparative trial with bromocriptine, cabergoline (at a dose of 0.5 mg twice weekly) was discontinued because of an adverse event in 4 of 221 patients (2%) while bromocriptine (at a dose of 2.5 mg two times a day) was discontinued in 14 of 231 patients (6%). The most common reasons for discontinuation from cabergoline were headache, nausea and vomiting (3, 2, and 2 patients, respectively); the most common reasons for discontinuation from bromocriptine were nausea, vomiting, headache, and dizziness or vertigo (10, 3, 3, and 3 patients, respectively). The incidence of the most common adverse events during the double-blind portion of the comparative trial with bromocriptine is presented in the following table.
|
*Reported at ≥1% for cabergoline |
||
|
Adverse
Event
*
|
Cabergoline
( n = 221 ) |
Bromocriptine
( n = 231 ) |
|
|
Number
(
percent
)
|
|
|
Gastrointestinal
|
|
|
| Nausea |
63 (29) |
100 (43) |
| Constipation |
15 (7) |
21 (9) |
| Abdominal pain |
12 (5) |
19 (8) |
| Dyspepsia |
11 (5) |
16 (7) |
| Vomiting |
9 (4) |
16 (7) |
| Dry mouth |
5 (2) |
2 (1) |
| Diarrhea |
4 (2) |
7 (3) |
| Flatulence |
4 (2) |
3 (1) |
| Throat irritation |
2 (1) |
0 |
| Toothache |
2 (1) |
0 |
|
Central
and
Peripheral
Nervous
System
|
|
|
| Headache |
58 (26) |
62 (27) |
| Dizziness |
38 (17) |
42 (18) |
| Vertigo |
9 (4) |
10 (4) |
| Paresthesia |
5 (2) |
6 (3) |
|
Body
As
A
Whole
|
|
|
| Asthenia |
13 (6) |
15 (6) |
| Fatigue |
10 (5) |
18 (8) |
| Syncope |
3 (1) |
3 (1) |
| Influenza-like symptoms |
2 (1) |
0 |
| Malaise |
2 (1) |
0 |
| Periorbital edema |
2 (1) |
2 (1) |
| Peripheral edema |
2 (1) |
1 |
|
Psychiatric
|
|
|
| Depression |
7 (3) |
5 (2) |
| Somnolence |
5 (2) |
5 (2) |
| Anorexia |
3 (1) |
3 (1) |
| Anxiety |
3 (1) |
3 (1) |
| Insomnia |
3 (1) |
2 (1) |
| Impaired concentration |
2 (1) |
1 |
| Nervousness |
2 (1) |
5 (2) |
|
Cardiovascular
|
|
|
| Hot flashes |
6 (3) |
3 (1) |
| Hypotension |
3 (1) |
4 (2) |
| Dependent edema |
2 (1) |
1 |
| Palpitation |
2 (1) |
5 (2) |
|
Reproductive
–
Female
|
|
|
| Breast pain |
5 (2) |
8 (3) |
| Dysmenorrhea |
2 (1) |
1 |
|
Skin
and
Appendages
|
|
|
| Acne |
3 (1) |
0 |
| Pruritus |
2 (1) |
1 |
|
Musculoskeletal
|
|
|
| Pain |
4 (2) |
6 (3) |
| Arthralgia |
2 (1) |
0 |
|
Respiratory
|
|
|
| Rhinitis |
2 (1) |
9 (4) |
|
Vision
|
|
|
| Abnormal Vision |
2 (1) |
2 (1) |
Other adverse events that were reported at an incidence of <1.0% in the overall clinical studies follow.
Body as a Whole: facial edema, influenza-like symptoms, malaise
Cardiovascular System: hypotension, syncope, palpitations
Digestive System: dry mouth, flatulence, diarrhea, anorexia
Metabolic and Nutritional System: weight loss, weight gain
Nervous System: somnolence, nervousness, paresthesia, insomnia, anxiety
Respiratory System: nasal stuffiness, epistaxis
Skin and Appendages: acne, pruritus
Special Senses: abnormal vision
Urogenital System: dysmenorrhea, increased libido
The safety of cabergoline has been evaluated in approximately 1,200 patients with Parkinson's disease in controlled and uncontrolled studies at dosages of up to 11.5 mg/day which greatly exceeds the maximum recommended dosage of cabergoline for hyperprolactinemic disorders. In addition to the adverse events that occurred in the patients with hyperprolactinemic disorders, the most common adverse events in patients with Parkinson's disease were dyskinesia, hallucinations, confusion, and peripheral edema. Heart failure, pleural effusion, pulmonary fibrosis, and gastric or duodenal ulcer occurred rarely. One case of constrictive pericarditis has been reported.
Drug Interactions (DRUG INTERACTIONS)
Cabergoline should not be administered concurrently with D2-antagonists, such as phenothiazines, butyrophenones, thioxanthenes, or metoclopramide.
How Supplied (HOW SUPPLIED)
Product: 50090-5834
NDC: 50090-5834-0 8 TABLET in a BOTTLE
Description (DESCRIPTION)
Cabergoline Tablets, USP contain Cabergoline USP a dopamine receptor agonist. The chemical name for Cabergoline USP is 1-[(6-allylergolin-8β-yl)-carbonyl]-1-[3-(dimethylamino)propyl]-3-ethylurea. Its molecular formula is C26H37N5O2, and its molecular weight is 451.62. The structural formula is as follows:
Cabergoline USP is a white powder soluble in ethyl alcohol, chloroform, and N, N-dimethylformamide (DMF); slightly soluble in 0.1N hydrochloric acid; very slightly soluble in n-hexane; and insoluble in water.
Cabergoline Tablets, USP for oral administration, contains 0.5 mg of Cabergoline USP. Inactive ingredients consist of microcrystalline cellulose, croscarmellose sodium, citric acid, and magnesium stearate.
Section 42229-5 (42229-5)
Clinical Studies: The prolactin-lowering efficacy of cabergoline was demonstrated in hyperprolactinemic women in two randomized, double-blind, comparative studies, one with placebo and the other with bromocriptine. In the placebo-controlled study (placebo n=20; cabergoline n=168), cabergoline produced a dose-related decrease in serum prolactin levels with prolactin normalized after 4 weeks of treatment in 29%, 76%, 74% and 95% of the patients receiving 0.125, 0.5, 0.75, and 1 mg twice weekly, respectively.
In the 8-week, double-blind period of the comparative trial with bromocriptine (cabergoline n=223; bromocriptine n=236 in the intent-to-treat analysis), prolactin was normalized in 77% of the patients treated with cabergoline at 0.5 mg twice weekly compared with 59% of those treated with bromocriptine at 2.5 mg twice daily. Restoration of menses occurred in 77% of the women treated with cabergoline, compared with 70% of those treated with bromocriptine. Among patients with galactorrhea, this symptom disappeared in 73% of those treated with cabergoline compared with 56% of those treated with bromocriptine.
Section 43679-0 (43679-0)
Mechanism of Action: The secretion of prolactin by the anterior pituitary is mainly under hypothalmic inhibitory control, likely exerted through release of dopamine by tuberoinfundibular neurons. Cabergoline is a long-acting dopamine receptor agonist with a high affinity for D2 receptors. Results of in vitro studies demonstrate that cabergoline exerts a direct inhibitory effect on the secretion of prolactin by rat pituitary lactotrophs. Cabergoline decreased serum prolactin levels in reserpinized rats. Receptor-binding studies indicate that cabergoline has low affinity for dopamine D1, α1- and α2-adrenergic, and 5-HT1- and 5-HT2-serotonin receptors.
Overdosage (OVERDOSAGE)
Overdosage might be expected to produce nasal congestion, syncope, or hallucinations. Measures to support blood pressure should be taken if necessary.
Cabergoline (CABERGOLINE)
Geriatric Use (GERIATRIC USE)
Clinical studies of cabergoline did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Pediatric Use (PEDIATRIC USE)
Safety and effectiveness of cabergoline in pediatric patients have not been established.
Nursing Mothers (NURSING MOTHERS)
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from cabergoline, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. Use of cabergoline for the inhibition or suppression of physiologic lactation is not recommended (see PRECAUTIONS section).
The prolactin-lowering action of cabergoline suggests that it will interfere with lactation. Due to this interference with lactation, cabergoline should not be given to women postpartum who are breastfeeding or who are planning to breastfeed.
Pharmacodynamics (PHARMACODYNAMICS)
Dose response with inhibition of plasma prolactin, onset of maximal effect, and duration of effect has been documented following single cabergoline doses to healthy volunteers (0.05 to 1.5 mg) and hyperprolactinemic patients (0.3 to 1 mg). In volunteers, prolactin inhibition was evident at doses >0.2 mg, while doses ≥0.5 mg caused maximal suppression in most subjects. Higher doses produce prolactin suppression in a greater proportion of subjects and with an earlier onset and longer duration of action. In 12 healthy volunteers, 0.5, 1 and 1.5 mg doses resulted in complete prolactin inhibition, with a maximum effect within 3 hours in 92% to 100% of subjects after the 1 and 1.5 mg doses compared with 50% of subjects after the 0.5 mg dose.
In hyperprolactinemic patients (n=51), the maximal prolactin decrease after a 0.6 mg single dose of cabergoline was comparable to 2.5 mg bromocriptine; however, the duration of effect was markedly longer (14 days vs. 24 hours). The time to maximal effect was shorter for bromocriptine than cabergoline (6 hours vs. 48 hours).
In 72 healthy volunteers, single or multiple doses (up to 2 mg) of cabergoline resulted in selective inhibition of prolactin with no apparent effect on other anterior pituitary hormones (GH, FSH, LH, ACTH, and TSH) or cortisol.
General Precautions
General: Initial doses higher than 1 mg may produce orthostatic hypotension. Care should be exercised when administering cabergoline with other medications known to lower blood pressure.
Food Drug Interaction (Food-Drug Interaction)
In 12 healthy adult volunteers, food did not alter cabergoline kinetics.
Information for Patients (Information for patients)
Patients should be altered to the possibility that patients may experience intense urges to spend money uncontrollably, intense urges to gamble, increased sexual urges, and other intense urges and the inability to control these urges while taking cabergoline. Advise patients to inform their healthcare provider if they develop new or increased uncontrolled spending, gambling urges, sexual urges, or other urges while being treated with cabergoline [See PRECAUTIONS].
Patients should be instructed to notify their physician if they suspect they are pregnant, become pregnant, or intend to become pregnant during therapy. A pregnancy test should be done if there is any suspicion of pregnancy and continuation of treatment should be discussed with their physician.
Patients should notify their physician if they develop shortness of breath, persistent cough, difficulty with breathing when lying down, or swelling in their extremities.
Pregnancy: Teratogenic Effects: Category B
Reproduction studies have been performed with cabergoline in mice, rats, and rabbits administered by gavage.
(Multiples of the maximum recommended human dose in this section are calculated on a body surface area basis using total mg/m2/week for animals and mg/m2/week for a 50 kg human.)
There were maternotoxic effects but no teratogenic effects in mice given cabergoline at doses up to 8 mg/kg/day (approximately 55 times the maximum recommended human dose) during the period of organogenesis.
A dose of 0.012 mg/kg/day (approximately 1/7 the maximum recommended human dose) during the period of organogenesis in rats caused an increase in post-implantation embryofetal losses. These losses could be due to the prolactin inhibitory properties of cabergoline in rats. At daily doses of 0.5 mg/kg/day (approximately 19 times the maximum recommended human dose) during the period of organogenesis in the rabbit, cabergoline caused maternotoxicity characterized by a loss of body weight and decreased food consumption. Doses of 4 mg/kg/day (approximately 150 times the maximum recommended human dose) during the period of organogenesis in the rabbit caused an increased occurrence of various malformations. However, in another study in rabbits, no treatment-related malformations or embryofetotoxicity were observed at doses up to 8 mg/kg/day (approximately 300 times the maximum recommended human dose).
In rats, doses higher than 0.003 mg/kg/day (approximately 1/28 the maximum recommended human dose) from 6 days before parturition and throughout the lactation period inhibited growth and caused death of offspring due to decreased milk secretion.
There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Carcinogenesis and Mutagenesis and Impairment of Fertility
Carcinogenicity studies were conducted in mice and rats with cabergoline given by gavage at doses up to 0.98 mg/kg/day and 0.32 mg/kg/day, respectively. These doses are 7 times and 4 times the maximum recommended human dose calculated on a body surface area basis using total mg/m2/week in rodents and mg/m2/week for a 50 kg human.
There was a slight increase in the incidence of cervical and uterine leiomyomas and uterine leiomyosarcomas in mice. In rats, there was a slight increase in malignant tumors of the cervix and uterus and interstitial cell adenomas. The occurrence of tumors in female rodents may be related to the prolonged suppression of prolactin secretion because prolactin is needed in rodents for the maintenance of the corpus luteum. In the absence of prolactin, the estrogen/progesterone ratio is increased, thereby increasing the risk for uterine tumors. In male rodents, the decrease in serum prolactin levels was associated with an increase in serum luteinizing hormone, which is thought to be a compensatory effect to maintain testicular steroid synthesis. Since these hormonal mechanisms are thought to be species-specific, the relevance of these tumors to humans is not known.
The mutagenic potential of cabergoline was evaluated and found to be negative in a battery of in vitro tests. These tests included the bacterial mutation (Ames) test with Salmonella typhimurium, the gene mutation assay with Schizosaccharomyces pombe P1 and V79 Chinese hamster cells, DNA damage and repair in Saccharomyces cerevisiae D4 , and chromosomal aberrations in human lymphocytes. Cabergoline was also negative in the bone marrow micronucleus test in the mouse.
In female rats, a daily dose of 0.003 mg/kg for 2 weeks prior to mating and throughout the mating period inhibited conception. This dose represents approximately 1/28 the maximum recommended human dose calculated on a body surface area basis using total mg/m2/week in rats and mg/m2/week for a 50 kg human.
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:38:07.024826 · Updated: 2026-03-14T21:53:19.065561