Zytiga
4e338e89-3cf2-48eb-b6e2-a06c608c6513
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
ZYTIGA is indicated in combination with prednisone for the treatment of patients with Metastatic castration-resistant prostate cancer (CRPC) Metastatic high-risk castration-sensitive prostate cancer (CSPC)
Dosage and Administration
Metastatic castration-resistant prostate cancer: ZYTIGA 1,000 mg orally once daily with prednisone 5 mg orally twice daily. ( 2.1 ) Metastatic castration-sensitive prostate cancer: ZYTIGA 1,000 mg orally once daily with prednisone 5 mg orally once daily. ( 2.2 ) Patients receiving ZYTIGA should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy. ZYTIGA tablets must be taken as a single dose once daily on an empty stomach. Do not eat food 2 hours before and 1 hour after taking ZYTIGA. The tablets must be swallowed whole with water. Do not crush or chew tablets. ( 2.3 ) Dose Modification: For patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the ZYTIGA starting dose to 250 mg once daily. ( 2.4 ) For patients who develop hepatotoxicity during treatment, hold ZYTIGA until recovery. Retreatment may be initiated at a reduced dose. ZYTIGA should be discontinued if patients develop severe hepatotoxicity. ( 2.4 )
Contraindications
None.
Warnings and Precautions
Mineralocorticoid excess: Closely monitor patients with cardiovascular disease. Control hypertension and correct hypokalemia before treatment. Monitor blood pressure, serum potassium and symptoms of fluid retention at least monthly. ( 5.1 ) Adrenocortical insufficiency: Monitor for symptoms and signs of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations. ( 5.2 ) Hepatotoxicity: Can be severe and fatal. Monitor liver function and modify, interrupt, or discontinue ZYTIGA dosing as recommended. ( 5.3 ) Increased fractures and mortality in combination with radium Ra 223 dichloride: Use of ZYTIGA plus prednisone/prednisolone in combination with radium Ra 223 dichloride is not recommended. ( 5.4 ) Embryo-Fetal Toxicity: ZYTIGA can cause fetal harm. Advise males with female partners of reproductive potential to use effective contraception. ( 5.5 , 8.1 , 8.3 ) Hypoglycemia: Severe hypoglycemia has been reported in patients with pre-existing diabetes who are taking medications containing thiazolidinediones (including pioglitazone) or repaglinide. Monitor blood glucose in patients with diabetes and assess if antidiabetic agent dose modifications are required. ( 5.6 )
Adverse Reactions
The following are discussed in more detail in other sections of the labeling: Hypokalemia, Fluid Retention, and Cardiovascular Adverse Reactions due to Mineralocorticoid Excess [see Warnings and Precautions (5.1) ] . Adrenocortical Insufficiency [see Warnings and Precautions (5.2) ] . Hepatotoxicity [see Warnings and Precautions (5.3) ] . Increased Fractures and Mortality in Combination with Radium Ra 223 Dichloride [see Warnings and Precautions (5.4) ].
Drug Interactions
CYP3A4 Inducers: Avoid concomitant strong CYP3A4 inducers during ZYTIGA treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency. ( 2.5 , 7.1 ) CYP2D6 Substrates: Avoid co-administration of ZYTIGA with CYP2D6 substrates that have a narrow therapeutic index. If an alternative treatment cannot be used, exercise caution and consider a dose reduction of the concomitant CYP2D6 substrate. ( 7.2 )
How Supplied
ZYTIGA ® (abiraterone acetate) Tablets are available in the strengths and packages listed below: ZYTIGA ® 500 mg film-coated Tablets Purple, oval-shaped tablets debossed with "AA" one side and "500" on the other side. NDC 57894-195-06, 60 tablets available in child-resistant high-density polyethylene bottles ZYTIGA ® 250 mg uncoated Tablets White to off-white, oval-shaped tablets debossed with "AA250" on one side. NDC 57894-150-12, 120 tablets available in child-resistant high-density polyethylene bottles
Storage and Handling
ZYTIGA ® (abiraterone acetate) Tablets are available in the strengths and packages listed below: ZYTIGA ® 500 mg film-coated Tablets Purple, oval-shaped tablets debossed with "AA" one side and "500" on the other side. NDC 57894-195-06, 60 tablets available in child-resistant high-density polyethylene bottles ZYTIGA ® 250 mg uncoated Tablets White to off-white, oval-shaped tablets debossed with "AA250" on one side. NDC 57894-150-12, 120 tablets available in child-resistant high-density polyethylene bottles
Description
ZYTIGA is indicated in combination with prednisone for the treatment of patients with Metastatic castration-resistant prostate cancer (CRPC) Metastatic high-risk castration-sensitive prostate cancer (CSPC)
Medication Information
Warnings and Precautions
Mineralocorticoid excess: Closely monitor patients with cardiovascular disease. Control hypertension and correct hypokalemia before treatment. Monitor blood pressure, serum potassium and symptoms of fluid retention at least monthly. ( 5.1 ) Adrenocortical insufficiency: Monitor for symptoms and signs of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations. ( 5.2 ) Hepatotoxicity: Can be severe and fatal. Monitor liver function and modify, interrupt, or discontinue ZYTIGA dosing as recommended. ( 5.3 ) Increased fractures and mortality in combination with radium Ra 223 dichloride: Use of ZYTIGA plus prednisone/prednisolone in combination with radium Ra 223 dichloride is not recommended. ( 5.4 ) Embryo-Fetal Toxicity: ZYTIGA can cause fetal harm. Advise males with female partners of reproductive potential to use effective contraception. ( 5.5 , 8.1 , 8.3 ) Hypoglycemia: Severe hypoglycemia has been reported in patients with pre-existing diabetes who are taking medications containing thiazolidinediones (including pioglitazone) or repaglinide. Monitor blood glucose in patients with diabetes and assess if antidiabetic agent dose modifications are required. ( 5.6 )
Indications and Usage
ZYTIGA is indicated in combination with prednisone for the treatment of patients with Metastatic castration-resistant prostate cancer (CRPC) Metastatic high-risk castration-sensitive prostate cancer (CSPC)
Dosage and Administration
Metastatic castration-resistant prostate cancer: ZYTIGA 1,000 mg orally once daily with prednisone 5 mg orally twice daily. ( 2.1 ) Metastatic castration-sensitive prostate cancer: ZYTIGA 1,000 mg orally once daily with prednisone 5 mg orally once daily. ( 2.2 ) Patients receiving ZYTIGA should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy. ZYTIGA tablets must be taken as a single dose once daily on an empty stomach. Do not eat food 2 hours before and 1 hour after taking ZYTIGA. The tablets must be swallowed whole with water. Do not crush or chew tablets. ( 2.3 ) Dose Modification: For patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the ZYTIGA starting dose to 250 mg once daily. ( 2.4 ) For patients who develop hepatotoxicity during treatment, hold ZYTIGA until recovery. Retreatment may be initiated at a reduced dose. ZYTIGA should be discontinued if patients develop severe hepatotoxicity. ( 2.4 )
Contraindications
None.
Adverse Reactions
The following are discussed in more detail in other sections of the labeling: Hypokalemia, Fluid Retention, and Cardiovascular Adverse Reactions due to Mineralocorticoid Excess [see Warnings and Precautions (5.1) ] . Adrenocortical Insufficiency [see Warnings and Precautions (5.2) ] . Hepatotoxicity [see Warnings and Precautions (5.3) ] . Increased Fractures and Mortality in Combination with Radium Ra 223 Dichloride [see Warnings and Precautions (5.4) ].
Drug Interactions
CYP3A4 Inducers: Avoid concomitant strong CYP3A4 inducers during ZYTIGA treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency. ( 2.5 , 7.1 ) CYP2D6 Substrates: Avoid co-administration of ZYTIGA with CYP2D6 substrates that have a narrow therapeutic index. If an alternative treatment cannot be used, exercise caution and consider a dose reduction of the concomitant CYP2D6 substrate. ( 7.2 )
Storage and Handling
ZYTIGA ® (abiraterone acetate) Tablets are available in the strengths and packages listed below: ZYTIGA ® 500 mg film-coated Tablets Purple, oval-shaped tablets debossed with "AA" one side and "500" on the other side. NDC 57894-195-06, 60 tablets available in child-resistant high-density polyethylene bottles ZYTIGA ® 250 mg uncoated Tablets White to off-white, oval-shaped tablets debossed with "AA250" on one side. NDC 57894-150-12, 120 tablets available in child-resistant high-density polyethylene bottles
How Supplied
ZYTIGA ® (abiraterone acetate) Tablets are available in the strengths and packages listed below: ZYTIGA ® 500 mg film-coated Tablets Purple, oval-shaped tablets debossed with "AA" one side and "500" on the other side. NDC 57894-195-06, 60 tablets available in child-resistant high-density polyethylene bottles ZYTIGA ® 250 mg uncoated Tablets White to off-white, oval-shaped tablets debossed with "AA250" on one side. NDC 57894-150-12, 120 tablets available in child-resistant high-density polyethylene bottles
Description
ZYTIGA is indicated in combination with prednisone for the treatment of patients with Metastatic castration-resistant prostate cancer (CRPC) Metastatic high-risk castration-sensitive prostate cancer (CSPC)
Section 42229-5
Hepatic Impairment
In patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the recommended dose of ZYTIGA to 250 mg once daily. In patients with moderate hepatic impairment monitor ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. If elevations in ALT and/or AST greater than 5 × upper limit of normal (ULN) or total bilirubin greater than 3 × ULN occur in patients with baseline moderate hepatic impairment, discontinue ZYTIGA and do not re-treat patients with ZYTIGA [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)] .
Do not use ZYTIGA in patients with baseline severe hepatic impairment (Child-Pugh Class C).
Section 42230-3
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 11/2024 | |
|
PATIENT INFORMATION
ZYTIGA ® ( Zye- tee- ga) (abiraterone acetate) tablets |
||
|
What is ZYTIGA?
ZYTIGA is a prescription medicine that is used along with prednisone. ZYTIGA is used to treat men with prostate cancer that has spread to other parts of the body. It is not known if ZYTIGA is safe and effective in females or children. |
||
Before taking ZYTIGA, tell your healthcare provider about all of your medical conditions, including if you:
You should not start or stop any medicine before you talk with the healthcare provider that prescribed ZYTIGA. Know the medicines you take. Keep a list of them with you to show to your healthcare provider and pharmacist when you get a new medicine. |
||
How should I take ZYTIGA?
|
||
|
What are the possible side effects of ZYTIGA?
ZYTIGA may cause serious side effects including:
|
||
|
|
|
|
||
|
|
|
| The most common side effects of ZYTIGA include: | ||
|
|
|
| ZYTIGA may cause fertility problems in males, which may affect the ability to father children. Talk to your healthcare provider if you have concerns about fertility.
These are not all the possible side effects of ZYTIGA. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store ZYTIGA?
|
||
|
General information about the safe and effective use of ZYTIGA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ZYTIGA for a condition for which it was not prescribed. Do not give ZYTIGA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ZYTIGA that is written for health professionals. |
||
|
What are the ingredients of ZYTIGA?
Active ingredient: abiraterone acetate Inactive ingredients: 500 mg film-coated tablets: colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, silicified microcrystalline cellulose, and sodium lauryl sulfate. The film-coating contains iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. 250 mg uncoated tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate. Manufactured for: Janssen Biotech, Inc., Horsham, PA 19044, USA For more information, call Janssen Biotech, Inc. at 1-800-526-7736 or go to www.Zytiga.com. For patent information: www.janssenpatents.com © Johnson & Johnson and its affiliates 2011, 2017, 2024 |
Section 44425-7
Storage and Handling
Store at 20 °C to 25 °C (68 °F to 77 °F); excursions permitted in the range from 15 °C to 30 °C (59 °F to 86 °F) [see USP Controlled Room Temperature].
Keep out of reach of children.
Based on its mechanism of action, ZYTIGA may harm a developing fetus. Women who are pregnant or women who may be pregnant should not handle ZYTIGA 250 mg uncoated tablets or other ZYTIGA tablets if broken, crushed, or damaged without protection, e.g., gloves [see Use in Specific Populations (8.1)] .
10 Overdosage
Human experience of overdose with ZYTIGA is limited.
There is no specific antidote. In the event of an overdose, stop ZYTIGA, undertake general supportive measures, including monitoring for arrhythmias and cardiac failure and assess liver function.
11 Description
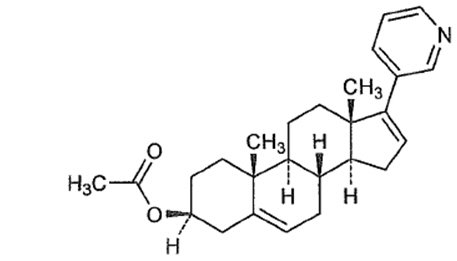
Abiraterone acetate, the active ingredient of ZYTIGA ® is the acetyl ester of abiraterone. Abiraterone is an inhibitor of CYP17 (17α-hydroxylase/C17,20-lyase). Each ZYTIGA tablet contains either 250 mg or 500 mg of abiraterone acetate. Abiraterone acetate is designated chemically as (3β)-17-(3-pyridinyl) androsta-5,16-dien-3-yl acetate and its structure is:
Abiraterone acetate is a white to off-white, non-hygroscopic, crystalline powder. Its molecular formula is C 26H 33NO 2 and it has a molecular weight of 391.55. Abiraterone acetate is a lipophilic compound with an octanol-water partition coefficient of 5.12 (Log P) and is practically insoluble in water. The pKa of the aromatic nitrogen is 5.19.
ZYTIGA tablets are available in 500 mg film-coated tablets and 250 mg uncoated tablets with the following inactive ingredients:
- 500 mg film-coated tablets: colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, silicified microcrystalline cellulose, and sodium lauryl sulfate. The coating, Opadry ®II Purple, contains iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
- 250 mg uncoated tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate.
5.6 Hypoglycemia
Severe hypoglycemia has been reported when ZYTIGA was administered to patients with pre-existing diabetes receiving medications containing thiazolidinediones (including pioglitazone) or repaglinide [see Drug Interactions (7.2)] . Monitor blood glucose in patients with diabetes during and after discontinuation of treatment with ZYTIGA. Assess if antidiabetic drug dosage needs to be adjusted to minimize the risk of hypoglycemia.
8.4 Pediatric Use
Safety and effectiveness of ZYTIGA in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of patients receiving ZYTIGA in randomized clinical trials, 70% of patients were 65 years and over and 27% were 75 years and over. No overall differences in safety or effectiveness were observed between these elderly patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
5.3 Hepatotoxicity
In postmarketing experience, there have been ZYTIGA-associated severe hepatic toxicity, including fulminant hepatitis, acute liver failure and deaths [see Adverse Reactions (6.2)] .
In the combined data of 5 randomized clinical trials, grade 3–4 ALT or AST increases (at least 5 × ULN) were reported in 6% of 2230 patients who received ZYTIGA, typically during the first 3 months after starting treatment. Patients whose baseline ALT or AST were elevated were more likely to experience liver test elevation than those beginning with normal values. Treatment discontinuation due to ALT and AST increases or abnormal hepatic function occurred in 1.1% of 2230 patients taking ZYTIGA. In these clinical trials, no deaths clearly related to ZYTIGA were reported due to hepatotoxicity events.
Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA, every two weeks for the first three months of treatment and monthly thereafter. In patients with baseline moderate hepatic impairment receiving a reduced ZYTIGA dose of 250 mg, measure ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient's baseline should prompt more frequent monitoring. If at any time AST or ALT rise above five times the ULN, or the bilirubin rises above three times the ULN, interrupt ZYTIGA treatment and closely monitor liver function.
Re-treatment with ZYTIGA at a reduced dose level may take place only after return of liver function tests to the patient's baseline or to AST and ALT less than or equal to 2.5 × ULN and total bilirubin less than or equal to 1.5 × ULN [see Dosage and Administration (2.4)] .
Permanently discontinue ZYTIGA for patients who develop a concurrent elevation of ALT greater than 3 × ULN and total bilirubin greater than 2 × ULN in the absence of biliary obstruction or other causes responsible for the concurrent elevation [see Dosage and Administration (2.4)] .
The safety of ZYTIGA re-treatment of patients who develop AST or ALT greater than or equal to 20 × ULN and/or bilirubin greater than or equal to 10 × ULN is unknown.
14 Clinical Studies
The efficacy and safety of ZYTIGA with prednisone was established in three randomized placebo-controlled international clinical studies. All patients in these studies received a GnRH analog or had prior bilateral orchiectomy. Patients with prior ketoconazole treatment for prostate cancer and a history of adrenal gland or pituitary disorders were excluded from these trials. Concurrent use of spironolactone was not allowed during the study period.
4 Contraindications
None.
6 Adverse Reactions
The following are discussed in more detail in other sections of the labeling:
- Hypokalemia, Fluid Retention, and Cardiovascular Adverse Reactions due to Mineralocorticoid Excess [see Warnings and Precautions (5.1)] .
- Adrenocortical Insufficiency [see Warnings and Precautions (5.2)] .
- Hepatotoxicity [see Warnings and Precautions (5.3)] .
- Increased Fractures and Mortality in Combination with Radium Ra 223 Dichloride [see Warnings and Precautions (5.4)].
7 Drug Interactions
- CYP3A4 Inducers: Avoid concomitant strong CYP3A4 inducers during ZYTIGA treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency. ( 2.5, 7.1)
- CYP2D6 Substrates: Avoid co-administration of ZYTIGA with CYP2D6 substrates that have a narrow therapeutic index. If an alternative treatment cannot be used, exercise caution and consider a dose reduction of the concomitant CYP2D6 substrate. ( 7.2)
12.3 Pharmacokinetics
Following administration of abiraterone acetate, the pharmacokinetics of abiraterone have been studied in healthy subjects and in patients with metastatic CRPC. In vivo, abiraterone acetate is converted to abiraterone. In clinical studies, abiraterone acetate plasma concentrations were below detectable levels (<0.2 ng/mL) in >99% of the analyzed samples.
1 Indications and Usage
ZYTIGA is indicated in combination with prednisone for the treatment of patients with
- Metastatic castration-resistant prostate cancer (CRPC)
- Metastatic high-risk castration-sensitive prostate cancer (CSPC)
12.1 Mechanism of Action
Abiraterone acetate (ZYTIGA) is converted in vivo to abiraterone, an androgen biosynthesis inhibitor, that inhibits 17 α-hydroxylase/C17,20-lyase (CYP17). This enzyme is expressed in testicular, adrenal, and prostatic tumor tissues and is required for androgen biosynthesis.
CYP17 catalyzes two sequential reactions: 1) the conversion of pregnenolone and progesterone to their 17α-hydroxy derivatives by 17α-hydroxylase activity and 2) the subsequent formation of dehydroepiandrosterone (DHEA) and androstenedione, respectively, by C17, 20 lyase activity. DHEA and androstenedione are androgens and are precursors of testosterone. Inhibition of CYP17 by abiraterone can also result in increased mineralocorticoid production by the adrenals [see Warnings and Precautions (5.1)] .
Androgen sensitive prostatic carcinoma responds to treatment that decreases androgen levels. Androgen deprivation therapies, such as treatment with GnRH agonists or orchiectomy, decrease androgen production in the testes but do not affect androgen production by the adrenals or in the tumor.
ZYTIGA decreased serum testosterone and other androgens in patients in the placebo-controlled clinical trial. It is not necessary to monitor the effect of ZYTIGA on serum testosterone levels.
Changes in serum prostate specific antigen (PSA) levels may be observed but have not been shown to correlate with clinical benefit in individual patients.
5.5 Embryo Fetal Toxicity
The safety and efficacy of ZYTIGA have not been established in females. Based on animal reproductive studies and mechanism of action, ZYTIGA can cause fetal harm and loss of pregnancy when administered to a pregnant female. In animal reproduction studies, oral administration of abiraterone acetate to pregnant rats during organogenesis caused adverse developmental effects at maternal exposures approximately ≥ 0.03 times the human exposure (AUC) at the recommended dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with ZYTIGA and for 3 weeks after the last dose of ZYTIGA [see Use in Specific Populations (8.1, 8.3)] . ZYTIGA should not be handled by females who are or may become pregnant [see How Supplied/Storage and Handling (16)].
5 Warnings and Precautions
- Mineralocorticoid excess: Closely monitor patients with cardiovascular disease. Control hypertension and correct hypokalemia before treatment. Monitor blood pressure, serum potassium and symptoms of fluid retention at least monthly. ( 5.1)
- Adrenocortical insufficiency: Monitor for symptoms and signs of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations. ( 5.2)
- Hepatotoxicity: Can be severe and fatal. Monitor liver function and modify, interrupt, or discontinue ZYTIGA dosing as recommended. ( 5.3)
- Increased fractures and mortality in combination with radium Ra 223 dichloride: Use of ZYTIGA plus prednisone/prednisolone in combination with radium Ra 223 dichloride is not recommended. ( 5.4)
- Embryo-Fetal Toxicity: ZYTIGA can cause fetal harm. Advise males with female partners of reproductive potential to use effective contraception. ( 5.5, 8.1, 8.3)
- Hypoglycemia: Severe hypoglycemia has been reported in patients with pre-existing diabetes who are taking medications containing thiazolidinediones (including pioglitazone) or repaglinide. Monitor blood glucose in patients with diabetes and assess if antidiabetic agent dose modifications are required. ( 5.6)
2 Dosage and Administration
Metastatic castration-resistant prostate cancer:
- ZYTIGA 1,000 mg orally once daily with prednisone 5 mg orally twicedaily. ( 2.1)
Metastatic castration-sensitive prostate cancer:
- ZYTIGA 1,000 mg orally once daily with prednisone 5 mg orally oncedaily. ( 2.2)
Patients receiving ZYTIGA should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy. ZYTIGA tablets must be taken as a single dose once daily on an empty stomach. Do not eat food 2 hours before and 1 hour after taking ZYTIGA. The tablets must be swallowed whole with water. Do not crush or chew tablets. ( 2.3)
Dose Modification:
- For patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the ZYTIGA starting dose to 250 mg once daily. ( 2.4)
- For patients who develop hepatotoxicity during treatment, hold ZYTIGA until recovery. Retreatment may be initiated at a reduced dose. ZYTIGA should be discontinued if patients develop severe hepatotoxicity. ( 2.4)
3 Dosage Forms and Strengths
Tablets (500 mg): purple, oval-shaped, film-coated tablets debossed with "AA" one side and "500" on the other side.
Tablets (250 mg): white to off-white, oval-shaped tablets debossed with "AA250" on one side.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during post approval use of ZYTIGA with prednisone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory, Thoracic and Mediastinal Disorders:non-infectious pneumonitis.
Musculoskeletal and Connective Tissue Disorders:myopathy, including rhabdomyolysis.
Hepatobiliary Disorders :fulminant hepatitis, including acute hepatic failure and death.
Cardiac Disorders:QT prolongation and Torsades de Pointes (observed in patients who developed hypokalemia or had underlying cardiovascular conditions).
Immune System Disorders – Hypersensitivity:anaphylactic reactions (severe allergic reactions that include, but are not limited to difficulty swallowing or breathing, swollen face, lips, tongue or throat, or an itchy rash (urticaria)).
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Two randomized placebo-controlled, multicenter clinical trials (COU-AA-301 and COU-AA-302) enrolled patients who had metastatic CRPC in which ZYTIGA was administered orally at a dose of 1,000 mg daily in combination with prednisone 5 mg twice daily in the active treatment arms. Placebo plus prednisone 5 mg twice daily was given to patients on the control arm. A third randomized placebo-controlled, multicenter clinical trial (LATITUDE) enrolled patients who had metastatic high-risk CSPC in which ZYTIGA was administered at a dose of 1,000 mg daily in combination with prednisone 5 mg once daily. Placebos were administered to patients in the control arm. Additionally, two other randomized, placebo-controlled trials were conducted in patients with metastatic CRPC. The safety data pooled from 2230 patients in the 5 randomized controlled trials constitute the basis for the data presented in the Warnings and Precautions, Grade 1–4 adverse reactions, and Grade 1–4 laboratory abnormalities. In all trials, a gonadotropin-releasing hormone (GnRH) analog or prior orchiectomy was required in both arms.
In the pooled data, median treatment duration was 11 months (0.1, 43) for ZYTIGA-treated patients and 7.2 months (0.1, 43) for placebo-treated patients. The most common adverse reactions (≥10%) that occurred more commonly (>2%) in the ZYTIGA arm were fatigue, arthralgia, hypertension, nausea, edema, hypokalemia, hot flush, diarrhea, vomiting, upper respiratory infection, cough, and headache. The most common laboratory abnormalities (>20%) that occurred more commonly (≥2%) in the ZYTIGA arm were anemia, elevated alkaline phosphatase, hypertriglyceridemia, lymphopenia, hypercholesterolemia, hyperglycemia, and hypokalemia. Grades 3–4 adverse events were reported for 53% of patients in the ZYTIGA arm and 46% of patients in the placebo arm. Treatment discontinuation was reported in 14% of patients in the ZYTIGA arm and 13% of patients in the placebo arm. The common adverse events (≥1%) resulting in discontinuation of ZYTIGA and prednisone were hepatotoxicity and cardiac disorders.
Deaths associated with treatment-emergent adverse events were reported for 7.5% of patients in the ZYTIGA arm and 6.6% of patients in the placebo arm. Of the patients in the ZYTIGA arm, the most common cause of death was disease progression (3.3%). Other reported causes of death in ≥5 patients included pneumonia, cardio-respiratory arrest, death (no additional information), and general physical health deterioration.
8 Use in Specific Populations
- Do not use ZYTIGA in patients with baseline severe hepatic impairment (Child-Pugh Class C). ( 8.6)
5.2 Adrenocortical Insufficiency
Adrenal insufficiency occurred in 0.3% of 2230 patients taking ZYTIGA and in 0.1% of 1763 patients taking placebo in the combined data of the 5 randomized, placebo-controlled clinical studies. Adrenocortical insufficiency was reported in patients receiving ZYTIGA in combination with prednisone, following interruption of daily steroids and/or with concurrent infection or stress.
Monitor patients for symptoms and signs of adrenocortical insufficiency, particularly if patients are withdrawn from prednisone, have prednisone dose reductions, or experience unusual stress. Symptoms and signs of adrenocortical insufficiency may be masked by adverse reactions associated with mineralocorticoid excess seen in patients treated with ZYTIGA. If clinically indicated, perform appropriate tests to confirm the diagnosis of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations [see Warnings and Precautions (5.1)].
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information)
8.7 Patients With Renal Impairment
No dosage adjustment is necessary for patients with renal impairment [see Clinical Pharmacology (12.3)] .
16 How Supplied/storage and Handling
ZYTIGA ® (abiraterone acetate) Tablets are available in the strengths and packages listed below:
-
ZYTIGA
® 500 mg film-coated Tablets
Purple, oval-shaped tablets debossed with "AA" one side and "500" on the other side.
NDC 57894-195-06, 60 tablets available in child-resistant high-density polyethylene bottles -
ZYTIGA
®
250 mg uncoated Tablets
White to off-white, oval-shaped tablets debossed with "AA250" on one side.
NDC 57894-150-12, 120 tablets available in child-resistant high-density polyethylene bottles
8.6 Patients With Hepatic Impairment
The pharmacokinetics of abiraterone were examined in subjects with baseline mild (N=8) or moderate (N=8) hepatic impairment (Child-Pugh Class A and B, respectively) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone after a single oral 1,000 mg dose of ZYTIGA increased by approximately 1.1-fold and 3.6-fold in subjects with mild and moderate baseline hepatic impairment, respectively compared to subjects with normal hepatic function.
In another trial, the pharmacokinetics of abiraterone were examined in subjects with baseline severe (N=8) hepatic impairment (Child-Pugh Class C) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone increased by approximately 7-fold and the fraction of free drug increased 2-fold in subjects with severe baseline hepatic impairment compared to subjects with normal hepatic function.
No dosage adjustment is necessary for patients with baseline mild hepatic impairment. In patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the recommended dose of ZYTIGA to 250 mg once daily. Do not use ZYTIGA in patients with baseline severe hepatic impairment (Child-Pugh Class C). If elevations in ALT or AST >5 × ULN or total bilirubin >3 × ULN occur in patients with baseline moderate hepatic impairment, discontinue ZYTIGA treatment [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)] .
For patients who develop hepatotoxicity during treatment, interruption of treatment and dosage adjustment may be required [see Dosage and Administration (2.4), Warnings and Precautions (5.3), and Clinical Pharmacology (12.3)].
2.1 Recommended Dose for Metastatic Crpc
The recommended dose of ZYTIGA is 1,000 mg (two 500 mg tablets or four 250 mg tablets) orally once daily with prednisone 5 mg orally twice daily.
2.3 Important Administration Instructions
Patients receiving ZYTIGA should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy.
ZYTIGA tablets must be taken as a single dose once daily on an empty stomach. Do not eat food 2 hours before and 1 hour after taking ZYTIGA. The tablets must be swallowed whole with water. Do not crush or chew tablets.
13.2 Animal Toxicology And/or Pharmacology
A dose-dependent increase in cataracts was observed in rats after daily oral abiraterone acetate administration for 26 weeks starting at ≥50 mg/kg/day (similar to the human clinical exposure based on AUC). In a 39-week monkey study with daily oral abiraterone acetate administration, no cataracts were observed at higher doses (2 times greater than the clinical exposure based on AUC).
7.1 Drugs That Inhibit Or Induce Cyp3a4 Enzymes
Based on in vitrodata, ZYTIGA is a substrate of CYP3A4.
In a dedicated drug interaction trial, co-administration of rifampin, a strong CYP3A4 inducer, decreased exposure of abiraterone by 55%. Avoid concomitant strong CYP3A4 inducers during ZYTIGA treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)] .
In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful effect on the pharmacokinetics of abiraterone [see Clinical Pharmacology (12.3)] .
2.2 Recommended Dose for Metastatic High Risk Cspc
The recommended dose of ZYTIGA is 1,000 mg (two 500 mg tablets or four 250 mg tablets) orally once daily with prednisone 5 mg administered orally once daily.
Principal Display Panel 250 Mg Tablet Bottle Label
NDC 57894-150-12
Rx only
Zytiga
®
(abiraterone acetate)
tablets
250 mg
Each tablet contains
250 mg of abiraterone acetate.
Warning: Women who are or
may be pregnant should not handle
ZYTIGA without gloves
(see Prescribing Information).
120 tablets
Principal Display Panel 500 Mg Tablet Bottle Label
NDC 57894-195-06
Zytiga
® 500 mg
(abiraterone acetate)
Tablets
Each film-coated tablet contains
500 mg of abiraterone acetate.
Rx only
60 film-coated tablets
7.2 Effects of Abiraterone On Drug Metabolizing Enzymes
ZYTIGA is an inhibitor of the hepatic drug-metabolizing enzymes CYP2D6 and CYP2C8. In a CYP2D6 drug-drug interaction trial, the C maxand AUC of dextromethorphan (CYP2D6 substrate) were increased 2.8- and 2.9-fold, respectively, when dextromethorphan was given with abiraterone acetate 1,000 mg daily and prednisone 5 mg twice daily. Avoid co-administration of abiraterone acetate with substrates of CYP2D6 with a narrow therapeutic index (e.g., thioridazine). If alternative treatments cannot be used, consider a dose reduction of the concomitant CYP2D6 substrate drug [see Clinical Pharmacology (12.3)] .
In a CYP2C8 drug-drug interaction trial in healthy subjects, the AUC of pioglitazone (CYP2C8 substrate) was increased by 46% when pioglitazone was given together with a single dose of 1,000 mg abiraterone acetate. Therefore, patients should be monitored closely for signs of toxicity related to a CYP2C8 substrate with a narrow therapeutic index if used concomitantly with ZYTIGA [see Clinical Pharmacology (12.3) and Warnings and Precautions (5.6)] .
2.5 Dose Modification Guidelines for Strong Cyp3a4 Inducers
Avoid concomitant strong CYP3A4 inducers (e.g., phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital) during ZYTIGA treatment.
If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency to twice a day only during the co-administration period (e.g., from 1,000 mg once daily to 1,000 mg twice a day). Reduce the dose back to the previous dose and frequency, if the concomitant strong CYP3A4 inducer is discontinued [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)] .
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
A two-year carcinogenicity study was conducted in rats at oral abiraterone acetate doses of 5, 15, and 50 mg/kg/day for males and 15, 50, and 150 mg/kg/day for females. Abiraterone acetate increased the combined incidence of interstitial cell adenomas and carcinomas in the testes at all dose levels tested. This finding is considered to be related to the pharmacological activity of abiraterone. Rats are regarded as more sensitive than humans to developing interstitial cell tumors in the testes. Abiraterone acetate was not carcinogenic in female rats at exposure levels up to 0.8 times the human clinical exposure based on AUC. Abiraterone acetate was not carcinogenic in a 6-month study in the transgenic (Tg.rasH2) mouse.
Abiraterone acetate and abiraterone was not mutagenic in an in vitro microbial mutagenesis (Ames) assay or clastogenic in an in vitro cytogenetic assay using primary human lymphocytes or an in vivo rat micronucleus assay.
In repeat-dose toxicity studies in male rats (13- and 26-weeks) and monkeys (39-weeks), atrophy, aspermia/hypospermia, and hyperplasia in the reproductive system were observed at ≥50 mg/kg/day in rats and ≥250 mg/kg/day in monkeys and were consistent with the antiandrogenic pharmacological activity of abiraterone. These effects were observed in rats at systemic exposures similar to humans and in monkeys at exposures approximately 0.6 times the AUC in humans.
In a fertility study in male rats, reduced organ weights of the reproductive system, sperm counts, sperm motility, altered sperm morphology and decreased fertility were observed in animals dosed for 4 weeks at ≥30 mg/kg/day orally. Mating of untreated females with males that received 30 mg/kg/day oral abiraterone acetate resulted in a reduced number of corpora lutea, implantations and live embryos and an increased incidence of pre-implantation loss. Effects on male rats were reversible after 16 weeks from the last abiraterone acetate administration.
In a fertility study in female rats, animals dosed orally for 2 weeks until day 7 of pregnancy at ≥30 mg/kg/day had an increased incidence of irregular or extended estrous cycles and pre-implantation loss (300 mg/kg/day). There were no differences in mating, fertility, and litter parameters in female rats that received abiraterone acetate. Effects on female rats were reversible after 4 weeks from the last abiraterone acetate administration.
The dose of 30 mg/kg/day in rats is approximately 0.3 times the recommended dose of 1,000 mg/day based on body surface area.
In 13- and 26-week studies in rats and 13- and 39-week studies in monkeys, a reduction in circulating testosterone levels occurred with abiraterone acetate at approximately one half the human clinical exposure based on AUC. As a result, decreases in organ weights and toxicities were observed in the male and female reproductive system, adrenal glands, liver, pituitary (rats only), and male mammary glands. The changes in the reproductive organs are consistent with the antiandrogenic pharmacological activity of abiraterone acetate.
5.4 Increased Fractures and Mortality in Combination With Radium Ra 223 Dichloride
ZYTIGA plus prednisone/prednisolone is not recommended for use in combination with radium Ra 223 dichloride outside of clinical trials.
The clinical efficacy and safety of concurrent initiation of ZYTIGA plus prednisone/prednisolone and radium Ra 223 dichloride was assessed in a randomized, placebo-controlled multicenter study (ERA-223 trial) in 806 patients with asymptomatic or mildly symptomatic castration-resistant prostate cancer with bone metastases. The study was unblinded early based on an Independent Data Monitoring Committee recommendation.
At the primary analysis, increased incidences of fractures (28.6% vs 11.4%) and deaths (38.5% vs 35.5%) have been observed in patients who received ZYTIGA plus prednisone/prednisolone in combination with radium Ra 223 dichloride compared to patients who received placebo in combination with ZYTIGA plus prednisone/prednisolone.
5.1 Hypokalemia, Fluid Retention, and Cardiovascular Adverse Reactions Due to Mineralocorticoid Excess
ZYTIGA may cause hypertension, hypokalemia, and fluid retention as a consequence of increased mineralocorticoid levels resulting from CYP17 inhibition [see Clinical Pharmacology (12.1)] . Monitor patients for hypertension, hypokalemia, and fluid retention at least once a month. Control hypertension and correct hypokalemia before and during treatment with ZYTIGA.
In the combined data from 4 placebo-controlled trials using prednisone 5 mg twice daily in combination with 1,000 mg abiraterone acetate daily, grades 3–4 hypokalemia were detected in 4% of patients on the ZYTIGA arm and 2% of patients on the placebo arm. Grades 3–4 hypertension were observed in 2% of patients each arm and grades 3–4 fluid retention in 1% of patients each arm.
In LATITUDE (a randomized placebo-controlled, multicenter clinical trial), which used prednisone 5 mg daily in combination with 1,000 mg abiraterone acetate daily, grades 3–4 hypokalemia were detected in 10% of patients on the ZYTIGA arm and 1% of patients on the placebo arm, grades 3–4 hypertension were observed in 20% of patients on the ZYTIGA arm and 10% of patients on the placebo arm. Grades 3–4 fluid retention occurred in 1% of patients each arm [see Adverse Reactions (6)] .
Closely monitor patients whose underlying medical conditions might be compromised by increases in blood pressure, hypokalemia or fluid retention, such as those with heart failure, recent myocardial infarction, cardiovascular disease, or ventricular arrhythmia. In postmarketing experience, QT prolongation and Torsades de Pointes have been observed in patients who develop hypokalemia while taking ZYTIGA.
The safety of ZYTIGA in patients with left ventricular ejection fraction <50% or New York Heart Association (NYHA) Class III or IV heart failure (in COU-AA-301) or NYHA Class II to IV heart failure (in COU-AA-302 and LATITUDE) has not been established because these patients were excluded from these randomized clinical trials [see Clinical Studies (14)] .
Structured Label Content
Section 42229-5 (42229-5)
Hepatic Impairment
In patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the recommended dose of ZYTIGA to 250 mg once daily. In patients with moderate hepatic impairment monitor ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. If elevations in ALT and/or AST greater than 5 × upper limit of normal (ULN) or total bilirubin greater than 3 × ULN occur in patients with baseline moderate hepatic impairment, discontinue ZYTIGA and do not re-treat patients with ZYTIGA [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)] .
Do not use ZYTIGA in patients with baseline severe hepatic impairment (Child-Pugh Class C).
Section 42230-3 (42230-3)
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 11/2024 | |
|
PATIENT INFORMATION
ZYTIGA ® ( Zye- tee- ga) (abiraterone acetate) tablets |
||
|
What is ZYTIGA?
ZYTIGA is a prescription medicine that is used along with prednisone. ZYTIGA is used to treat men with prostate cancer that has spread to other parts of the body. It is not known if ZYTIGA is safe and effective in females or children. |
||
Before taking ZYTIGA, tell your healthcare provider about all of your medical conditions, including if you:
You should not start or stop any medicine before you talk with the healthcare provider that prescribed ZYTIGA. Know the medicines you take. Keep a list of them with you to show to your healthcare provider and pharmacist when you get a new medicine. |
||
How should I take ZYTIGA?
|
||
|
What are the possible side effects of ZYTIGA?
ZYTIGA may cause serious side effects including:
|
||
|
|
|
|
||
|
|
|
| The most common side effects of ZYTIGA include: | ||
|
|
|
| ZYTIGA may cause fertility problems in males, which may affect the ability to father children. Talk to your healthcare provider if you have concerns about fertility.
These are not all the possible side effects of ZYTIGA. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store ZYTIGA?
|
||
|
General information about the safe and effective use of ZYTIGA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ZYTIGA for a condition for which it was not prescribed. Do not give ZYTIGA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ZYTIGA that is written for health professionals. |
||
|
What are the ingredients of ZYTIGA?
Active ingredient: abiraterone acetate Inactive ingredients: 500 mg film-coated tablets: colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, silicified microcrystalline cellulose, and sodium lauryl sulfate. The film-coating contains iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. 250 mg uncoated tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate. Manufactured for: Janssen Biotech, Inc., Horsham, PA 19044, USA For more information, call Janssen Biotech, Inc. at 1-800-526-7736 or go to www.Zytiga.com. For patent information: www.janssenpatents.com © Johnson & Johnson and its affiliates 2011, 2017, 2024 |
Section 44425-7 (44425-7)
Storage and Handling
Store at 20 °C to 25 °C (68 °F to 77 °F); excursions permitted in the range from 15 °C to 30 °C (59 °F to 86 °F) [see USP Controlled Room Temperature].
Keep out of reach of children.
Based on its mechanism of action, ZYTIGA may harm a developing fetus. Women who are pregnant or women who may be pregnant should not handle ZYTIGA 250 mg uncoated tablets or other ZYTIGA tablets if broken, crushed, or damaged without protection, e.g., gloves [see Use in Specific Populations (8.1)] .
10 Overdosage (10 OVERDOSAGE)
Human experience of overdose with ZYTIGA is limited.
There is no specific antidote. In the event of an overdose, stop ZYTIGA, undertake general supportive measures, including monitoring for arrhythmias and cardiac failure and assess liver function.
11 Description (11 DESCRIPTION)
Abiraterone acetate, the active ingredient of ZYTIGA ® is the acetyl ester of abiraterone. Abiraterone is an inhibitor of CYP17 (17α-hydroxylase/C17,20-lyase). Each ZYTIGA tablet contains either 250 mg or 500 mg of abiraterone acetate. Abiraterone acetate is designated chemically as (3β)-17-(3-pyridinyl) androsta-5,16-dien-3-yl acetate and its structure is:
Abiraterone acetate is a white to off-white, non-hygroscopic, crystalline powder. Its molecular formula is C 26H 33NO 2 and it has a molecular weight of 391.55. Abiraterone acetate is a lipophilic compound with an octanol-water partition coefficient of 5.12 (Log P) and is practically insoluble in water. The pKa of the aromatic nitrogen is 5.19.
ZYTIGA tablets are available in 500 mg film-coated tablets and 250 mg uncoated tablets with the following inactive ingredients:
- 500 mg film-coated tablets: colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, silicified microcrystalline cellulose, and sodium lauryl sulfate. The coating, Opadry ®II Purple, contains iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
- 250 mg uncoated tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate.
5.6 Hypoglycemia
Severe hypoglycemia has been reported when ZYTIGA was administered to patients with pre-existing diabetes receiving medications containing thiazolidinediones (including pioglitazone) or repaglinide [see Drug Interactions (7.2)] . Monitor blood glucose in patients with diabetes during and after discontinuation of treatment with ZYTIGA. Assess if antidiabetic drug dosage needs to be adjusted to minimize the risk of hypoglycemia.
8.4 Pediatric Use
Safety and effectiveness of ZYTIGA in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of patients receiving ZYTIGA in randomized clinical trials, 70% of patients were 65 years and over and 27% were 75 years and over. No overall differences in safety or effectiveness were observed between these elderly patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
5.3 Hepatotoxicity
In postmarketing experience, there have been ZYTIGA-associated severe hepatic toxicity, including fulminant hepatitis, acute liver failure and deaths [see Adverse Reactions (6.2)] .
In the combined data of 5 randomized clinical trials, grade 3–4 ALT or AST increases (at least 5 × ULN) were reported in 6% of 2230 patients who received ZYTIGA, typically during the first 3 months after starting treatment. Patients whose baseline ALT or AST were elevated were more likely to experience liver test elevation than those beginning with normal values. Treatment discontinuation due to ALT and AST increases or abnormal hepatic function occurred in 1.1% of 2230 patients taking ZYTIGA. In these clinical trials, no deaths clearly related to ZYTIGA were reported due to hepatotoxicity events.
Measure serum transaminases (ALT and AST) and bilirubin levels prior to starting treatment with ZYTIGA, every two weeks for the first three months of treatment and monthly thereafter. In patients with baseline moderate hepatic impairment receiving a reduced ZYTIGA dose of 250 mg, measure ALT, AST, and bilirubin prior to the start of treatment, every week for the first month, every two weeks for the following two months of treatment and monthly thereafter. Promptly measure serum total bilirubin, AST, and ALT if clinical symptoms or signs suggestive of hepatotoxicity develop. Elevations of AST, ALT, or bilirubin from the patient's baseline should prompt more frequent monitoring. If at any time AST or ALT rise above five times the ULN, or the bilirubin rises above three times the ULN, interrupt ZYTIGA treatment and closely monitor liver function.
Re-treatment with ZYTIGA at a reduced dose level may take place only after return of liver function tests to the patient's baseline or to AST and ALT less than or equal to 2.5 × ULN and total bilirubin less than or equal to 1.5 × ULN [see Dosage and Administration (2.4)] .
Permanently discontinue ZYTIGA for patients who develop a concurrent elevation of ALT greater than 3 × ULN and total bilirubin greater than 2 × ULN in the absence of biliary obstruction or other causes responsible for the concurrent elevation [see Dosage and Administration (2.4)] .
The safety of ZYTIGA re-treatment of patients who develop AST or ALT greater than or equal to 20 × ULN and/or bilirubin greater than or equal to 10 × ULN is unknown.
14 Clinical Studies (14 CLINICAL STUDIES)
The efficacy and safety of ZYTIGA with prednisone was established in three randomized placebo-controlled international clinical studies. All patients in these studies received a GnRH analog or had prior bilateral orchiectomy. Patients with prior ketoconazole treatment for prostate cancer and a history of adrenal gland or pituitary disorders were excluded from these trials. Concurrent use of spironolactone was not allowed during the study period.
4 Contraindications (4 CONTRAINDICATIONS)
None.
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following are discussed in more detail in other sections of the labeling:
- Hypokalemia, Fluid Retention, and Cardiovascular Adverse Reactions due to Mineralocorticoid Excess [see Warnings and Precautions (5.1)] .
- Adrenocortical Insufficiency [see Warnings and Precautions (5.2)] .
- Hepatotoxicity [see Warnings and Precautions (5.3)] .
- Increased Fractures and Mortality in Combination with Radium Ra 223 Dichloride [see Warnings and Precautions (5.4)].
7 Drug Interactions (7 DRUG INTERACTIONS)
- CYP3A4 Inducers: Avoid concomitant strong CYP3A4 inducers during ZYTIGA treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency. ( 2.5, 7.1)
- CYP2D6 Substrates: Avoid co-administration of ZYTIGA with CYP2D6 substrates that have a narrow therapeutic index. If an alternative treatment cannot be used, exercise caution and consider a dose reduction of the concomitant CYP2D6 substrate. ( 7.2)
12.3 Pharmacokinetics
Following administration of abiraterone acetate, the pharmacokinetics of abiraterone have been studied in healthy subjects and in patients with metastatic CRPC. In vivo, abiraterone acetate is converted to abiraterone. In clinical studies, abiraterone acetate plasma concentrations were below detectable levels (<0.2 ng/mL) in >99% of the analyzed samples.
1 Indications and Usage (1 INDICATIONS AND USAGE)
ZYTIGA is indicated in combination with prednisone for the treatment of patients with
- Metastatic castration-resistant prostate cancer (CRPC)
- Metastatic high-risk castration-sensitive prostate cancer (CSPC)
12.1 Mechanism of Action
Abiraterone acetate (ZYTIGA) is converted in vivo to abiraterone, an androgen biosynthesis inhibitor, that inhibits 17 α-hydroxylase/C17,20-lyase (CYP17). This enzyme is expressed in testicular, adrenal, and prostatic tumor tissues and is required for androgen biosynthesis.
CYP17 catalyzes two sequential reactions: 1) the conversion of pregnenolone and progesterone to their 17α-hydroxy derivatives by 17α-hydroxylase activity and 2) the subsequent formation of dehydroepiandrosterone (DHEA) and androstenedione, respectively, by C17, 20 lyase activity. DHEA and androstenedione are androgens and are precursors of testosterone. Inhibition of CYP17 by abiraterone can also result in increased mineralocorticoid production by the adrenals [see Warnings and Precautions (5.1)] .
Androgen sensitive prostatic carcinoma responds to treatment that decreases androgen levels. Androgen deprivation therapies, such as treatment with GnRH agonists or orchiectomy, decrease androgen production in the testes but do not affect androgen production by the adrenals or in the tumor.
ZYTIGA decreased serum testosterone and other androgens in patients in the placebo-controlled clinical trial. It is not necessary to monitor the effect of ZYTIGA on serum testosterone levels.
Changes in serum prostate specific antigen (PSA) levels may be observed but have not been shown to correlate with clinical benefit in individual patients.
5.5 Embryo Fetal Toxicity (5.5 Embryo-Fetal Toxicity)
The safety and efficacy of ZYTIGA have not been established in females. Based on animal reproductive studies and mechanism of action, ZYTIGA can cause fetal harm and loss of pregnancy when administered to a pregnant female. In animal reproduction studies, oral administration of abiraterone acetate to pregnant rats during organogenesis caused adverse developmental effects at maternal exposures approximately ≥ 0.03 times the human exposure (AUC) at the recommended dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with ZYTIGA and for 3 weeks after the last dose of ZYTIGA [see Use in Specific Populations (8.1, 8.3)] . ZYTIGA should not be handled by females who are or may become pregnant [see How Supplied/Storage and Handling (16)].
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Mineralocorticoid excess: Closely monitor patients with cardiovascular disease. Control hypertension and correct hypokalemia before treatment. Monitor blood pressure, serum potassium and symptoms of fluid retention at least monthly. ( 5.1)
- Adrenocortical insufficiency: Monitor for symptoms and signs of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations. ( 5.2)
- Hepatotoxicity: Can be severe and fatal. Monitor liver function and modify, interrupt, or discontinue ZYTIGA dosing as recommended. ( 5.3)
- Increased fractures and mortality in combination with radium Ra 223 dichloride: Use of ZYTIGA plus prednisone/prednisolone in combination with radium Ra 223 dichloride is not recommended. ( 5.4)
- Embryo-Fetal Toxicity: ZYTIGA can cause fetal harm. Advise males with female partners of reproductive potential to use effective contraception. ( 5.5, 8.1, 8.3)
- Hypoglycemia: Severe hypoglycemia has been reported in patients with pre-existing diabetes who are taking medications containing thiazolidinediones (including pioglitazone) or repaglinide. Monitor blood glucose in patients with diabetes and assess if antidiabetic agent dose modifications are required. ( 5.6)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
Metastatic castration-resistant prostate cancer:
- ZYTIGA 1,000 mg orally once daily with prednisone 5 mg orally twicedaily. ( 2.1)
Metastatic castration-sensitive prostate cancer:
- ZYTIGA 1,000 mg orally once daily with prednisone 5 mg orally oncedaily. ( 2.2)
Patients receiving ZYTIGA should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy. ZYTIGA tablets must be taken as a single dose once daily on an empty stomach. Do not eat food 2 hours before and 1 hour after taking ZYTIGA. The tablets must be swallowed whole with water. Do not crush or chew tablets. ( 2.3)
Dose Modification:
- For patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the ZYTIGA starting dose to 250 mg once daily. ( 2.4)
- For patients who develop hepatotoxicity during treatment, hold ZYTIGA until recovery. Retreatment may be initiated at a reduced dose. ZYTIGA should be discontinued if patients develop severe hepatotoxicity. ( 2.4)
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Tablets (500 mg): purple, oval-shaped, film-coated tablets debossed with "AA" one side and "500" on the other side.
Tablets (250 mg): white to off-white, oval-shaped tablets debossed with "AA250" on one side.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during post approval use of ZYTIGA with prednisone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory, Thoracic and Mediastinal Disorders:non-infectious pneumonitis.
Musculoskeletal and Connective Tissue Disorders:myopathy, including rhabdomyolysis.
Hepatobiliary Disorders :fulminant hepatitis, including acute hepatic failure and death.
Cardiac Disorders:QT prolongation and Torsades de Pointes (observed in patients who developed hypokalemia or had underlying cardiovascular conditions).
Immune System Disorders – Hypersensitivity:anaphylactic reactions (severe allergic reactions that include, but are not limited to difficulty swallowing or breathing, swollen face, lips, tongue or throat, or an itchy rash (urticaria)).
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Two randomized placebo-controlled, multicenter clinical trials (COU-AA-301 and COU-AA-302) enrolled patients who had metastatic CRPC in which ZYTIGA was administered orally at a dose of 1,000 mg daily in combination with prednisone 5 mg twice daily in the active treatment arms. Placebo plus prednisone 5 mg twice daily was given to patients on the control arm. A third randomized placebo-controlled, multicenter clinical trial (LATITUDE) enrolled patients who had metastatic high-risk CSPC in which ZYTIGA was administered at a dose of 1,000 mg daily in combination with prednisone 5 mg once daily. Placebos were administered to patients in the control arm. Additionally, two other randomized, placebo-controlled trials were conducted in patients with metastatic CRPC. The safety data pooled from 2230 patients in the 5 randomized controlled trials constitute the basis for the data presented in the Warnings and Precautions, Grade 1–4 adverse reactions, and Grade 1–4 laboratory abnormalities. In all trials, a gonadotropin-releasing hormone (GnRH) analog or prior orchiectomy was required in both arms.
In the pooled data, median treatment duration was 11 months (0.1, 43) for ZYTIGA-treated patients and 7.2 months (0.1, 43) for placebo-treated patients. The most common adverse reactions (≥10%) that occurred more commonly (>2%) in the ZYTIGA arm were fatigue, arthralgia, hypertension, nausea, edema, hypokalemia, hot flush, diarrhea, vomiting, upper respiratory infection, cough, and headache. The most common laboratory abnormalities (>20%) that occurred more commonly (≥2%) in the ZYTIGA arm were anemia, elevated alkaline phosphatase, hypertriglyceridemia, lymphopenia, hypercholesterolemia, hyperglycemia, and hypokalemia. Grades 3–4 adverse events were reported for 53% of patients in the ZYTIGA arm and 46% of patients in the placebo arm. Treatment discontinuation was reported in 14% of patients in the ZYTIGA arm and 13% of patients in the placebo arm. The common adverse events (≥1%) resulting in discontinuation of ZYTIGA and prednisone were hepatotoxicity and cardiac disorders.
Deaths associated with treatment-emergent adverse events were reported for 7.5% of patients in the ZYTIGA arm and 6.6% of patients in the placebo arm. Of the patients in the ZYTIGA arm, the most common cause of death was disease progression (3.3%). Other reported causes of death in ≥5 patients included pneumonia, cardio-respiratory arrest, death (no additional information), and general physical health deterioration.
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
- Do not use ZYTIGA in patients with baseline severe hepatic impairment (Child-Pugh Class C). ( 8.6)
5.2 Adrenocortical Insufficiency
Adrenal insufficiency occurred in 0.3% of 2230 patients taking ZYTIGA and in 0.1% of 1763 patients taking placebo in the combined data of the 5 randomized, placebo-controlled clinical studies. Adrenocortical insufficiency was reported in patients receiving ZYTIGA in combination with prednisone, following interruption of daily steroids and/or with concurrent infection or stress.
Monitor patients for symptoms and signs of adrenocortical insufficiency, particularly if patients are withdrawn from prednisone, have prednisone dose reductions, or experience unusual stress. Symptoms and signs of adrenocortical insufficiency may be masked by adverse reactions associated with mineralocorticoid excess seen in patients treated with ZYTIGA. If clinically indicated, perform appropriate tests to confirm the diagnosis of adrenocortical insufficiency. Increased dosage of corticosteroids may be indicated before, during and after stressful situations [see Warnings and Precautions (5.1)].
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA-approved patient labeling (Patient Information)
8.7 Patients With Renal Impairment (8.7 Patients with Renal Impairment)
No dosage adjustment is necessary for patients with renal impairment [see Clinical Pharmacology (12.3)] .
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
ZYTIGA ® (abiraterone acetate) Tablets are available in the strengths and packages listed below:
-
ZYTIGA
® 500 mg film-coated Tablets
Purple, oval-shaped tablets debossed with "AA" one side and "500" on the other side.
NDC 57894-195-06, 60 tablets available in child-resistant high-density polyethylene bottles -
ZYTIGA
®
250 mg uncoated Tablets
White to off-white, oval-shaped tablets debossed with "AA250" on one side.
NDC 57894-150-12, 120 tablets available in child-resistant high-density polyethylene bottles
8.6 Patients With Hepatic Impairment (8.6 Patients with Hepatic Impairment)
The pharmacokinetics of abiraterone were examined in subjects with baseline mild (N=8) or moderate (N=8) hepatic impairment (Child-Pugh Class A and B, respectively) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone after a single oral 1,000 mg dose of ZYTIGA increased by approximately 1.1-fold and 3.6-fold in subjects with mild and moderate baseline hepatic impairment, respectively compared to subjects with normal hepatic function.
In another trial, the pharmacokinetics of abiraterone were examined in subjects with baseline severe (N=8) hepatic impairment (Child-Pugh Class C) and in 8 healthy control subjects with normal hepatic function. The systemic exposure (AUC) of abiraterone increased by approximately 7-fold and the fraction of free drug increased 2-fold in subjects with severe baseline hepatic impairment compared to subjects with normal hepatic function.
No dosage adjustment is necessary for patients with baseline mild hepatic impairment. In patients with baseline moderate hepatic impairment (Child-Pugh Class B), reduce the recommended dose of ZYTIGA to 250 mg once daily. Do not use ZYTIGA in patients with baseline severe hepatic impairment (Child-Pugh Class C). If elevations in ALT or AST >5 × ULN or total bilirubin >3 × ULN occur in patients with baseline moderate hepatic impairment, discontinue ZYTIGA treatment [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)] .
For patients who develop hepatotoxicity during treatment, interruption of treatment and dosage adjustment may be required [see Dosage and Administration (2.4), Warnings and Precautions (5.3), and Clinical Pharmacology (12.3)].
2.1 Recommended Dose for Metastatic Crpc (2.1 Recommended Dose for Metastatic CRPC)
The recommended dose of ZYTIGA is 1,000 mg (two 500 mg tablets or four 250 mg tablets) orally once daily with prednisone 5 mg orally twice daily.
2.3 Important Administration Instructions
Patients receiving ZYTIGA should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy.
ZYTIGA tablets must be taken as a single dose once daily on an empty stomach. Do not eat food 2 hours before and 1 hour after taking ZYTIGA. The tablets must be swallowed whole with water. Do not crush or chew tablets.
13.2 Animal Toxicology And/or Pharmacology (13.2 Animal Toxicology and/or Pharmacology)
A dose-dependent increase in cataracts was observed in rats after daily oral abiraterone acetate administration for 26 weeks starting at ≥50 mg/kg/day (similar to the human clinical exposure based on AUC). In a 39-week monkey study with daily oral abiraterone acetate administration, no cataracts were observed at higher doses (2 times greater than the clinical exposure based on AUC).
7.1 Drugs That Inhibit Or Induce Cyp3a4 Enzymes (7.1 Drugs that Inhibit or Induce CYP3A4 Enzymes)
Based on in vitrodata, ZYTIGA is a substrate of CYP3A4.
In a dedicated drug interaction trial, co-administration of rifampin, a strong CYP3A4 inducer, decreased exposure of abiraterone by 55%. Avoid concomitant strong CYP3A4 inducers during ZYTIGA treatment. If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)] .
In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful effect on the pharmacokinetics of abiraterone [see Clinical Pharmacology (12.3)] .
2.2 Recommended Dose for Metastatic High Risk Cspc (2.2 Recommended Dose for Metastatic High-risk CSPC)
The recommended dose of ZYTIGA is 1,000 mg (two 500 mg tablets or four 250 mg tablets) orally once daily with prednisone 5 mg administered orally once daily.
Principal Display Panel 250 Mg Tablet Bottle Label (PRINCIPAL DISPLAY PANEL - 250 mg Tablet Bottle Label)
NDC 57894-150-12
Rx only
Zytiga
®
(abiraterone acetate)
tablets
250 mg
Each tablet contains
250 mg of abiraterone acetate.
Warning: Women who are or
may be pregnant should not handle
ZYTIGA without gloves
(see Prescribing Information).
120 tablets
Principal Display Panel 500 Mg Tablet Bottle Label (PRINCIPAL DISPLAY PANEL - 500 mg Tablet Bottle Label)
NDC 57894-195-06
Zytiga
® 500 mg
(abiraterone acetate)
Tablets
Each film-coated tablet contains
500 mg of abiraterone acetate.
Rx only
60 film-coated tablets
7.2 Effects of Abiraterone On Drug Metabolizing Enzymes (7.2 Effects of Abiraterone on Drug Metabolizing Enzymes)
ZYTIGA is an inhibitor of the hepatic drug-metabolizing enzymes CYP2D6 and CYP2C8. In a CYP2D6 drug-drug interaction trial, the C maxand AUC of dextromethorphan (CYP2D6 substrate) were increased 2.8- and 2.9-fold, respectively, when dextromethorphan was given with abiraterone acetate 1,000 mg daily and prednisone 5 mg twice daily. Avoid co-administration of abiraterone acetate with substrates of CYP2D6 with a narrow therapeutic index (e.g., thioridazine). If alternative treatments cannot be used, consider a dose reduction of the concomitant CYP2D6 substrate drug [see Clinical Pharmacology (12.3)] .
In a CYP2C8 drug-drug interaction trial in healthy subjects, the AUC of pioglitazone (CYP2C8 substrate) was increased by 46% when pioglitazone was given together with a single dose of 1,000 mg abiraterone acetate. Therefore, patients should be monitored closely for signs of toxicity related to a CYP2C8 substrate with a narrow therapeutic index if used concomitantly with ZYTIGA [see Clinical Pharmacology (12.3) and Warnings and Precautions (5.6)] .
2.5 Dose Modification Guidelines for Strong Cyp3a4 Inducers (2.5 Dose Modification Guidelines for Strong CYP3A4 Inducers)
Avoid concomitant strong CYP3A4 inducers (e.g., phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital) during ZYTIGA treatment.
If a strong CYP3A4 inducer must be co-administered, increase the ZYTIGA dosing frequency to twice a day only during the co-administration period (e.g., from 1,000 mg once daily to 1,000 mg twice a day). Reduce the dose back to the previous dose and frequency, if the concomitant strong CYP3A4 inducer is discontinued [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)] .
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
A two-year carcinogenicity study was conducted in rats at oral abiraterone acetate doses of 5, 15, and 50 mg/kg/day for males and 15, 50, and 150 mg/kg/day for females. Abiraterone acetate increased the combined incidence of interstitial cell adenomas and carcinomas in the testes at all dose levels tested. This finding is considered to be related to the pharmacological activity of abiraterone. Rats are regarded as more sensitive than humans to developing interstitial cell tumors in the testes. Abiraterone acetate was not carcinogenic in female rats at exposure levels up to 0.8 times the human clinical exposure based on AUC. Abiraterone acetate was not carcinogenic in a 6-month study in the transgenic (Tg.rasH2) mouse.
Abiraterone acetate and abiraterone was not mutagenic in an in vitro microbial mutagenesis (Ames) assay or clastogenic in an in vitro cytogenetic assay using primary human lymphocytes or an in vivo rat micronucleus assay.
In repeat-dose toxicity studies in male rats (13- and 26-weeks) and monkeys (39-weeks), atrophy, aspermia/hypospermia, and hyperplasia in the reproductive system were observed at ≥50 mg/kg/day in rats and ≥250 mg/kg/day in monkeys and were consistent with the antiandrogenic pharmacological activity of abiraterone. These effects were observed in rats at systemic exposures similar to humans and in monkeys at exposures approximately 0.6 times the AUC in humans.
In a fertility study in male rats, reduced organ weights of the reproductive system, sperm counts, sperm motility, altered sperm morphology and decreased fertility were observed in animals dosed for 4 weeks at ≥30 mg/kg/day orally. Mating of untreated females with males that received 30 mg/kg/day oral abiraterone acetate resulted in a reduced number of corpora lutea, implantations and live embryos and an increased incidence of pre-implantation loss. Effects on male rats were reversible after 16 weeks from the last abiraterone acetate administration.
In a fertility study in female rats, animals dosed orally for 2 weeks until day 7 of pregnancy at ≥30 mg/kg/day had an increased incidence of irregular or extended estrous cycles and pre-implantation loss (300 mg/kg/day). There were no differences in mating, fertility, and litter parameters in female rats that received abiraterone acetate. Effects on female rats were reversible after 4 weeks from the last abiraterone acetate administration.
The dose of 30 mg/kg/day in rats is approximately 0.3 times the recommended dose of 1,000 mg/day based on body surface area.
In 13- and 26-week studies in rats and 13- and 39-week studies in monkeys, a reduction in circulating testosterone levels occurred with abiraterone acetate at approximately one half the human clinical exposure based on AUC. As a result, decreases in organ weights and toxicities were observed in the male and female reproductive system, adrenal glands, liver, pituitary (rats only), and male mammary glands. The changes in the reproductive organs are consistent with the antiandrogenic pharmacological activity of abiraterone acetate.
5.4 Increased Fractures and Mortality in Combination With Radium Ra 223 Dichloride (5.4 Increased Fractures and Mortality in Combination with Radium Ra 223 Dichloride)
ZYTIGA plus prednisone/prednisolone is not recommended for use in combination with radium Ra 223 dichloride outside of clinical trials.
The clinical efficacy and safety of concurrent initiation of ZYTIGA plus prednisone/prednisolone and radium Ra 223 dichloride was assessed in a randomized, placebo-controlled multicenter study (ERA-223 trial) in 806 patients with asymptomatic or mildly symptomatic castration-resistant prostate cancer with bone metastases. The study was unblinded early based on an Independent Data Monitoring Committee recommendation.
At the primary analysis, increased incidences of fractures (28.6% vs 11.4%) and deaths (38.5% vs 35.5%) have been observed in patients who received ZYTIGA plus prednisone/prednisolone in combination with radium Ra 223 dichloride compared to patients who received placebo in combination with ZYTIGA plus prednisone/prednisolone.
5.1 Hypokalemia, Fluid Retention, and Cardiovascular Adverse Reactions Due to Mineralocorticoid Excess (5.1 Hypokalemia, Fluid Retention, and Cardiovascular Adverse Reactions due to Mineralocorticoid Excess)
ZYTIGA may cause hypertension, hypokalemia, and fluid retention as a consequence of increased mineralocorticoid levels resulting from CYP17 inhibition [see Clinical Pharmacology (12.1)] . Monitor patients for hypertension, hypokalemia, and fluid retention at least once a month. Control hypertension and correct hypokalemia before and during treatment with ZYTIGA.
In the combined data from 4 placebo-controlled trials using prednisone 5 mg twice daily in combination with 1,000 mg abiraterone acetate daily, grades 3–4 hypokalemia were detected in 4% of patients on the ZYTIGA arm and 2% of patients on the placebo arm. Grades 3–4 hypertension were observed in 2% of patients each arm and grades 3–4 fluid retention in 1% of patients each arm.
In LATITUDE (a randomized placebo-controlled, multicenter clinical trial), which used prednisone 5 mg daily in combination with 1,000 mg abiraterone acetate daily, grades 3–4 hypokalemia were detected in 10% of patients on the ZYTIGA arm and 1% of patients on the placebo arm, grades 3–4 hypertension were observed in 20% of patients on the ZYTIGA arm and 10% of patients on the placebo arm. Grades 3–4 fluid retention occurred in 1% of patients each arm [see Adverse Reactions (6)] .
Closely monitor patients whose underlying medical conditions might be compromised by increases in blood pressure, hypokalemia or fluid retention, such as those with heart failure, recent myocardial infarction, cardiovascular disease, or ventricular arrhythmia. In postmarketing experience, QT prolongation and Torsades de Pointes have been observed in patients who develop hypokalemia while taking ZYTIGA.
The safety of ZYTIGA in patients with left ventricular ejection fraction <50% or New York Heart Association (NYHA) Class III or IV heart failure (in COU-AA-301) or NYHA Class II to IV heart failure (in COU-AA-302 and LATITUDE) has not been established because these patients were excluded from these randomized clinical trials [see Clinical Studies (14)] .
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:46:22.692614 · Updated: 2026-03-14T22:24:42.571850