Valproate Sodium Injection
3dfb6da1-f66a-4618-9f74-d2d6c16c0b58
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
Valproate sodium injection is indicated as an intravenous alternative in patients in whom oral administration of valproate products is temporarily not feasible in the following conditions: • Monotherapy and adjunctive therapy of complex partial seizures and simple and complex absence seizures; adjunctive therapy in patients with multiple seizure types that include absence seizures ( 1 )
Dosage and Administration
Valproate sodium injection is intended for intravenous use only. • Epilepsy o Complex Partial Seizures in Adults and Children 10 years of age or older: Initial dose is 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response. Maximum recommended dose is 60 mg/kg/day ( 2.1 ). o Simple and Complex Absence Seizures: Initial dose is 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response. Maximum recommended dose is 60 mg/kg/day ( 2.1 ).
Contraindications
• Valproate sodium injection should not be administered to patients with hepatic disease or significant hepatic dysfunction [see Warnings and Precautions ( 5.1 )] . • Valproate sodium injection is contraindicated in patients known to have mitochondrial disorders caused by mutations in mitochondrial DNA polymerase γ (POLG; e.g., Alpers-Huttenlocher Syndrome) and children under two years of age who are suspected of having a POLG-related disorder [see Warnings and Precautions ( 5.1 )] . • Valproate sodium injection is contraindicated in patients with known hypersensitivity to the drug [see Warnings and Precautions ( 5.11 )] . • Valproate sodium injection is contraindicated in patients with known urea cycle disorders [see Warnings and Precautions ( 5.6 )] . • For use in prophylaxis of migraine headaches: Valproate sodium injection is contraindicated in women who are pregnant and in women of childbearing potential who are not using effective contraception [see Warnings and Precautions ( 5.2 , 5.3 , 5.4 ) and Use in Specific Populations ( 8.1 )] .
Warnings and Precautions
• Hepatotoxicity; evaluate high risk populations and monitor serum liver tests ( 5.1 ) • Birth defects, decreased IQ, and neurodevelopmental disorders following in utero exposure; should not be used to treat women with epilepsy or bipolar disorder who are pregnant or who plan to become pregnant or to treat a woman of childbearing potential unless other medications have failed to provide adequate symptom control or are otherwise unacceptable ( 5.2 , 5.3 , 5.4 ) • Pancreatitis; valproate sodium should ordinarily be discontinued ( 5.5 ) • Bleeding and other hematopoietic disorders; monitor platelet counts and coagulation tests ( 5.7 ) • Hyperammonemia and hyperammonemic encephalopathy; measure ammonia level if unexplained lethargy and vomiting or changes in mental status, and also with concomitant topiramate use; consider discontinuation of valproate therapy ( 5.6 , 5.8 , 5.9 ) • Hypothermia; Hypothermia has been reported during valproate therapy with or without associated hyperammonemia. This adverse reaction can also occur in patients using concomitant topiramate ( 5.10 ) • Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan hypersensitivity reaction; discontinue valproate sodium ( 5.11 ) • Somnolence in the elderly can occur. Valproate sodium dosage should be increased slowly and with regular monitoring for fluid and nutritional intake ( 5.13 )
Adverse Reactions
WARNING: LIFE THREATENING ADVERSE REACTIONS See full prescribing information for complete boxed warning. • Hepatotoxicity, including fatalities, usually during the first 6 months of treatment. Children under the age of two years and patients with mitochondrial disorders are at higher risk. Monitor patients closely, and perform serum liver testing prior to therapy and at frequent intervals thereafter ( 5.1 ) • Fetal Risk, particularly neural tube defects, other major malformations, and decreased IQ ( 5.2 , 5.3 , 5.4 ) • Pancreatitis, including fatal hemorrhagic cases ( 5.5 )
Drug Interactions
• Hepatic enzyme-inducing drugs (e.g., phenytoin, carbamazepine, phenobarbital, primidone, rifampin) can increase valproate clearance, while enzyme inhibitors (e.g., felbamate) can decrease valproate clearance. Therefore increased monitoring of valproate and concomitant drug concentrations and dosage adjustment are indicated whenever enzyme-inducing or inhibiting drugs are introduced or withdrawn ( 7.1 ) • Aspirin, carbapenem antibiotics, estrogen-containing hormonal contraceptives: Monitoring of valproate concentrations is recommended ( 7.1 ) • Co-administration of valproate can affect the pharmacokinetics of other drugs (e.g. diazepam, ethosuximide, lamotrigine, phenytoin) by inhibiting their metabolism or protein binding displacement ( 7.2 ) • Patients stabilized on rufinamide should begin valproate therapy at a low dose, and titrate to clinically effective dose ( 7.2 ) • Dosage adjustment of amitriptyline/nortriptyline, propofol, warfarin, and zidovudine may be necessary if used concomitantly with valproate ( 7.2 ) • Topiramate: Hyperammonemia and encephalopathy ( 5.9 , 7.3 )
How Supplied
Valproate Sodium Injection, equivalent to 100 mg of valproic acid per mL, is a clear, colorless solution supplied as: Manufacturer Product Number Unit of Sale Strength Each Unit PRX439405 NDC 63323-494-16 Available in trays of 10 vials. 500 mg per 5 mL (100 mg per mL) NDC 63323-494-41 5 mL Single-dose Vial Store at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature]. Preservative Free. Unused portion of container should be discarded. The container closure is not made with natural rubber latex.
Storage and Handling
Valproate Sodium Injection, equivalent to 100 mg of valproic acid per mL, is a clear, colorless solution supplied as: Manufacturer Product Number Unit of Sale Strength Each Unit PRX439405 NDC 63323-494-16 Available in trays of 10 vials. 500 mg per 5 mL (100 mg per mL) NDC 63323-494-41 5 mL Single-dose Vial Store at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature]. Preservative Free. Unused portion of container should be discarded. The container closure is not made with natural rubber latex.
Description
WARNING: LIFE THREATENING ADVERSE REACTIONS See full prescribing information for complete boxed warning. • Hepatotoxicity, including fatalities, usually during the first 6 months of treatment. Children under the age of two years and patients with mitochondrial disorders are at higher risk. Monitor patients closely, and perform serum liver testing prior to therapy and at frequent intervals thereafter ( 5.1 ) • Fetal Risk, particularly neural tube defects, other major malformations, and decreased IQ ( 5.2 , 5.3 , 5.4 ) • Pancreatitis, including fatal hemorrhagic cases ( 5.5 )
Medication Information
Warnings and Precautions
• Hepatotoxicity; evaluate high risk populations and monitor serum liver tests ( 5.1 ) • Birth defects, decreased IQ, and neurodevelopmental disorders following in utero exposure; should not be used to treat women with epilepsy or bipolar disorder who are pregnant or who plan to become pregnant or to treat a woman of childbearing potential unless other medications have failed to provide adequate symptom control or are otherwise unacceptable ( 5.2 , 5.3 , 5.4 ) • Pancreatitis; valproate sodium should ordinarily be discontinued ( 5.5 ) • Bleeding and other hematopoietic disorders; monitor platelet counts and coagulation tests ( 5.7 ) • Hyperammonemia and hyperammonemic encephalopathy; measure ammonia level if unexplained lethargy and vomiting or changes in mental status, and also with concomitant topiramate use; consider discontinuation of valproate therapy ( 5.6 , 5.8 , 5.9 ) • Hypothermia; Hypothermia has been reported during valproate therapy with or without associated hyperammonemia. This adverse reaction can also occur in patients using concomitant topiramate ( 5.10 ) • Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan hypersensitivity reaction; discontinue valproate sodium ( 5.11 ) • Somnolence in the elderly can occur. Valproate sodium dosage should be increased slowly and with regular monitoring for fluid and nutritional intake ( 5.13 )
Indications and Usage
Valproate sodium injection is indicated as an intravenous alternative in patients in whom oral administration of valproate products is temporarily not feasible in the following conditions: • Monotherapy and adjunctive therapy of complex partial seizures and simple and complex absence seizures; adjunctive therapy in patients with multiple seizure types that include absence seizures ( 1 )
Dosage and Administration
Valproate sodium injection is intended for intravenous use only. • Epilepsy o Complex Partial Seizures in Adults and Children 10 years of age or older: Initial dose is 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response. Maximum recommended dose is 60 mg/kg/day ( 2.1 ). o Simple and Complex Absence Seizures: Initial dose is 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response. Maximum recommended dose is 60 mg/kg/day ( 2.1 ).
Contraindications
• Valproate sodium injection should not be administered to patients with hepatic disease or significant hepatic dysfunction [see Warnings and Precautions ( 5.1 )] . • Valproate sodium injection is contraindicated in patients known to have mitochondrial disorders caused by mutations in mitochondrial DNA polymerase γ (POLG; e.g., Alpers-Huttenlocher Syndrome) and children under two years of age who are suspected of having a POLG-related disorder [see Warnings and Precautions ( 5.1 )] . • Valproate sodium injection is contraindicated in patients with known hypersensitivity to the drug [see Warnings and Precautions ( 5.11 )] . • Valproate sodium injection is contraindicated in patients with known urea cycle disorders [see Warnings and Precautions ( 5.6 )] . • For use in prophylaxis of migraine headaches: Valproate sodium injection is contraindicated in women who are pregnant and in women of childbearing potential who are not using effective contraception [see Warnings and Precautions ( 5.2 , 5.3 , 5.4 ) and Use in Specific Populations ( 8.1 )] .
Adverse Reactions
WARNING: LIFE THREATENING ADVERSE REACTIONS See full prescribing information for complete boxed warning. • Hepatotoxicity, including fatalities, usually during the first 6 months of treatment. Children under the age of two years and patients with mitochondrial disorders are at higher risk. Monitor patients closely, and perform serum liver testing prior to therapy and at frequent intervals thereafter ( 5.1 ) • Fetal Risk, particularly neural tube defects, other major malformations, and decreased IQ ( 5.2 , 5.3 , 5.4 ) • Pancreatitis, including fatal hemorrhagic cases ( 5.5 )
Drug Interactions
• Hepatic enzyme-inducing drugs (e.g., phenytoin, carbamazepine, phenobarbital, primidone, rifampin) can increase valproate clearance, while enzyme inhibitors (e.g., felbamate) can decrease valproate clearance. Therefore increased monitoring of valproate and concomitant drug concentrations and dosage adjustment are indicated whenever enzyme-inducing or inhibiting drugs are introduced or withdrawn ( 7.1 ) • Aspirin, carbapenem antibiotics, estrogen-containing hormonal contraceptives: Monitoring of valproate concentrations is recommended ( 7.1 ) • Co-administration of valproate can affect the pharmacokinetics of other drugs (e.g. diazepam, ethosuximide, lamotrigine, phenytoin) by inhibiting their metabolism or protein binding displacement ( 7.2 ) • Patients stabilized on rufinamide should begin valproate therapy at a low dose, and titrate to clinically effective dose ( 7.2 ) • Dosage adjustment of amitriptyline/nortriptyline, propofol, warfarin, and zidovudine may be necessary if used concomitantly with valproate ( 7.2 ) • Topiramate: Hyperammonemia and encephalopathy ( 5.9 , 7.3 )
Storage and Handling
Valproate Sodium Injection, equivalent to 100 mg of valproic acid per mL, is a clear, colorless solution supplied as: Manufacturer Product Number Unit of Sale Strength Each Unit PRX439405 NDC 63323-494-16 Available in trays of 10 vials. 500 mg per 5 mL (100 mg per mL) NDC 63323-494-41 5 mL Single-dose Vial Store at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature]. Preservative Free. Unused portion of container should be discarded. The container closure is not made with natural rubber latex.
How Supplied
Valproate Sodium Injection, equivalent to 100 mg of valproic acid per mL, is a clear, colorless solution supplied as: Manufacturer Product Number Unit of Sale Strength Each Unit PRX439405 NDC 63323-494-16 Available in trays of 10 vials. 500 mg per 5 mL (100 mg per mL) NDC 63323-494-41 5 mL Single-dose Vial Store at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature]. Preservative Free. Unused portion of container should be discarded. The container closure is not made with natural rubber latex.
Description
WARNING: LIFE THREATENING ADVERSE REACTIONS See full prescribing information for complete boxed warning. • Hepatotoxicity, including fatalities, usually during the first 6 months of treatment. Children under the age of two years and patients with mitochondrial disorders are at higher risk. Monitor patients closely, and perform serum liver testing prior to therapy and at frequent intervals thereafter ( 5.1 ) • Fetal Risk, particularly neural tube defects, other major malformations, and decreased IQ ( 5.2 , 5.3 , 5.4 ) • Pancreatitis, including fatal hemorrhagic cases ( 5.5 )
Data
Human
Neural tube defects and other structural abnormalities
There is an extensive body of evidence demonstrating that exposure to valproate in utero increases the risk of neural tube defects and other structural abnormalities. Based on published data from the CDC's National Birth Defects Prevention Network, the risk of spina bifida in the general population is about 0.06 to 0.07% (6 to 7 in 10,000 births) compared to the risk following in utero valproate exposure estimated to be approximately 1 to 2% (100 to 200 in 10,000 births).
The NAAED Pregnancy Registry has reported a major malformation rate of 9-11% in the offspring of women exposed to an average of 1,000 mg/day of valproate monotherapy during pregnancy. These data show an up to a five-fold increased risk for any major malformation following valproate exposure in utero compared to the risk following exposure in utero to other AEDs taken as monotherapy. The major congenital malformations included cases of neural tube defects, cardiovascular malformations, craniofacial defects (e.g., oral clefts, craniosynostosis), hypospadias, limb malformations (e.g., clubfoot, polydactyly), and other malformations of varying severity involving other body systems [see Warnings and Precautions (5.2)].
Effect on IQ and neurodevelopmental effects
Published epidemiological studies have indicated that children exposed to valproate in utero have lower IQ scores than children exposed to either another AED in utero or to no AEDs in utero. The largest of these studies1 is a prospective cohort study conducted in the United States and United Kingdom that found that children with prenatal exposure to valproate (n=62) had lower IQ scores at age 6 (97 [95% C.I. 94-101]) than children with prenatal exposure to the other anti-epileptic drug monotherapy treatments evaluated: lamotrigine (108 [95% C.I. 105–110]), carbamazepine (105 [95% C.I. 102–108]) and phenytoin (108 [95% C.I. 104–112]). It is not known when during pregnancy cognitive effects in valproate-exposed children occur. Because the women in this study were exposed to AEDs throughout pregnancy, whether the risk for decreased IQ was related to a particular time period during pregnancy could not be assessed [see Warnings and Precautions (5.3)].
Although the available studies have methodological limitations, the weight of the evidence supports a causal association between valproate exposure in utero and subsequent adverse effects on neurodevelopment, including increases in autism spectrum disorders and attention deficit/hyperactivity disorder (ADHD). An observational study has suggested that exposure to valproate products during pregnancy increases the risk of autism spectrum disorders. In this study, children born to mothers who had used valproate products during pregnancy had 2.9 times the risk (95% confidence interval [CI]: 1.7-4.9) of developing autism spectrum disorders compared to children born to mothers not exposed to valproate products during pregnancy. The absolute risks for autism spectrum disorders were 4.4% (95% CI: 2.6%-7.5%) in valproate-exposed children and 1.5% (95% CI: 1.5%-1.6%) in children not exposed to valproate products. Another observational study found that children who were exposed to valproate in utero had an increased risk of ADHD (adjusted HR 1.48; 95% CI, 1.09-2.00) compared with the unexposed children. Because these studies were observational in nature, conclusions regarding a causal association between in utero valproate exposure and an increased risk of autism spectrum disorder and ADHD cannot be considered definitive.
Other
There are published case reports of fatal hepatic failure in offspring of women who used valproate during pregnancy.
Animal
In developmental toxicity studies conducted in mice, rats, rabbits, and monkeys, increased rates of fetal structural abnormalities, intrauterine growth retardation, and embryo- fetal death occurred following administration of valproate to pregnant animals during organogenesis at clinically relevant doses (calculated on a body surface area [mg/m2] basis). Valproate induced malformations of multiple organ systems, including skeletal, cardiac, and urogenital defects. In mice, in addition to other malformations, fetal neural tube defects have been reported following valproate administration during critical periods of organogenesis, and the teratogenic response correlated with peak maternal drug levels. Behavioral abnormalities (including cognitive, locomotor, and social interaction deficits) and brain histopathological changes have also been reported in mice and rat offspring exposed prenatally to clinically relevant doses of valproate.
Section 42229-5
Hepatotoxicity
6.2 Mania
Although valproate sodium has not been evaluated for safety and efficacy in the treatment of manic episodes associated with bipolar disorder, the following adverse reactions not listed above were reported by 1% or more of patients from two placebo-controlled clinical trials of divalproex sodium tablets.
Body as a Whole: Chills, neck pain, neck rigidity.
Cardiovascular System: Hypotension, postural hypotension, vasodilation.
Digestive System: Fecal incontinence, gastroenteritis, glossitis.
Musculoskeletal System: Arthrosis.
Nervous System: Agitation, catatonic reaction, hypokinesia, reflexes increased, tardive dyskinesia, vertigo.
Skin and Appendages: Furunculosis, maculopapular rash, seborrhea.
Special Senses: Conjunctivitis, dry eyes, eye pain.
Urogenital: Dysuria.
1.1 Epilepsy
Valproate sodium injection is indicated as an intravenous alternative in patients for whom oral administration of valproate products is temporarily not feasible in the following conditions:
Valproate sodium injection is indicated as monotherapy and adjunctive therapy in the treatment of patients with complex partial seizures that occur either in isolation or in association with other types of seizures. Valproate sodium injection is also indicated for use as sole and adjunctive therapy in the treatment of patients with simple and complex absence seizures, and adjunctively in patients with multiple seizure types that include absence seizures.
Simple absence is defined as very brief clouding of the sensorium or loss of consciousness accompanied by certain generalized epileptic discharges without other detectable clinical signs. Complex absence is the term used when other signs are also present.
See Warnings and Precautions (5.1) for statement regarding fatal hepatic dysfunction.
2.1 Epilepsy
Valproate sodium injection is for intravenous use only.
Use of valproate sodium injection for periods of more than 14 days has not been studied. Patients should be switched to oral valproate products as soon as it is clinically feasible.
Valproate sodium injection should be administered as a 60 minute infusion (but not more than 20 mg/min) with the same frequency as the oral products, although plasma concentration monitoring and dosage adjustments may be necessary.
In one clinical safety study, approximately 90 patients with epilepsy and with no measurable plasma levels of valproate were given single infusions of valproate sodium injection (up to 15 mg/kg and mean dose of 1,184 mg) over 5 to 10 minutes (1.5 to 3 mg/kg/min). Patients generally tolerated the more rapid infusions well [see Adverse Reactions (6.1)]. This study was not designed to assess the effectiveness of these regimens. For pharmacokinetics with rapid infusions, see Clinical Pharmacology (12.3).
6.1 Epilepsy
The data described in the following section were obtained using Depakote (divalproex sodium) tablets.
Based on a placebo-controlled trial of adjunctive therapy for treatment of complex partial seizures, divalproex sodium was generally well tolerated with most adverse reactions rated as mild to moderate in severity. Intolerance was the primary reason for discontinuation in the divalproex sodium-treated patients (6%), compared to 1% of placebo-treated patients.
Table 2 lists treatment-emergent adverse reactions which were reported by ≥ 5% of divalproex sodium-treated patients and for which the incidence was greater than in the placebo group, in the placebo-controlled trial of adjunctive therapy for treatment of complex partial seizures. Since patients were also treated with other antiepilepsy drugs, it is not possible, in most cases, to determine whether the following adverse reactions can be ascribed to divalproex sodium alone, or the combination of divalproex sodium and other antiepilepsy drugs.
|
Body System/Reaction |
Divalproex Sodium %
|
Placebo %
|
|
Body as a Whole |
||
|
Headache |
31 |
21 |
|
Asthenia |
27 |
7 |
|
Fever |
6 |
4 |
|
Gastrointestinal System |
||
|
Nausea |
48 |
14 |
|
Vomiting |
27 |
7 |
|
Abdominal Pain |
23 |
6 |
|
Diarrhea |
13 |
6 |
|
Anorexia |
12 |
0 |
|
Dyspepsia |
8 |
4 |
|
Constipation |
5 |
1 |
|
Nervous System |
||
|
Somnolence |
27 |
11 |
|
Tremor |
25 |
6 |
|
Dizziness |
25 |
13 |
|
Diplopia |
16 |
9 |
|
Amblyopia/Blurred Vision |
12 |
9 |
|
Ataxia |
8 |
1 |
|
Nystagmus |
8 |
1 |
|
Emotional Lability |
6 |
4 |
|
Thinking Abnormal |
6 |
0 |
|
Amnesia |
5 |
1 |
|
Respiratory System |
||
|
Flu Syndrome |
12 |
9 |
|
Infection |
12 |
6 |
|
Bronchitis |
5 |
1 |
|
Rhinitis |
5 |
4 |
|
Other |
||
|
Alopecia |
6 |
1 |
|
Weight Loss |
6 |
0 |
Table 3 lists treatment-emergent adverse reactions which were reported by ≥ 5% of patients in the high dose valproate group, and for which the incidence was greater than in the low dose group, in a controlled trial of divalproex sodium monotherapy treatment of complex partial seizures. Since patients were being titrated off another antiepilepsy drug during the first portion of the trial, it is not possible, in many cases, to determine whether the following adverse reactions can be ascribed to divalproex sodium alone, or the combination of valproate and other antiepilepsy drugs.
|
Body System/Reaction |
High Dose %
|
Low Dose %
|
|
Body as a Whole |
||
|
Asthenia |
21 |
10 |
|
Nausea |
34 |
26 |
|
Diarrhea |
23 |
19 |
|
Vomiting |
23 |
15 |
|
Abdominal Pain |
12 |
9 |
|
Anorexia |
11 |
4 |
|
Dyspepsia |
11 |
10 |
|
Thrombocytopenia |
24 |
1 |
|
Ecchymosis |
5 |
4 |
|
Weight Gain |
9 |
4 |
|
Peripheral Edema |
8 |
3 |
|
Tremor |
57 |
19 |
|
Somnolence |
30 |
18 |
|
Dizziness |
18 |
13 |
|
Insomnia |
15 |
9 |
|
Nervousness |
11 |
7 |
|
Amnesia |
7 |
4 |
|
Nystagmus |
7 |
1 |
|
Depression |
5 |
4 |
|
Infection |
20 |
13 |
|
Pharyngitis |
8 |
2 |
|
Dyspnea |
5 |
1 |
|
Alopecia |
24 |
13 |
|
Special Senses |
||
|
Amblyopia/Blurred Vision |
8 |
4 |
|
Tinnitus |
7 |
1 |
1 Headache was the only adverse reaction that occurred in ≥ 5% of patients in the high dose group and at an equal or greater incidence in the low dose group.
The following additional adverse reactions were reported by greater than 1% but less than 5% of the 358 patients treated with valproate in the controlled trials of complex partial seizures:
Body as a Whole: Back pain, chest pain, malaise.
Cardiovascular System: Tachycardia, hypertension, palpitation.
Digestive System: Increased appetite, flatulence, hematemesis, eructation, pancreatitis, periodontal abscess.
Hemic and Lymphatic System: Petechia.
Metabolic and Nutritional Disorders: SGOT increased, SGPT increased.
Musculoskeletal System: Myalgia, twitching, arthralgia, leg cramps, myasthenia.
Nervous System: Anxiety, confusion, abnormal gait, paresthesia, hypertonia, incoordination, abnormal dreams, personality disorder.
Respiratory System: Sinusitis, cough increased, pneumonia, epistaxis.
Skin and Appendages: Rash, pruritus, dry skin.
Special Senses: Taste perversion, abnormal vision, deafness, otitis media.
Urogenital System: Urinary incontinence, vaginitis, dysmenorrhea, amenorrhea, urinary frequency.
6.3 Migraine
Although valproate has not been evaluated for safety and efficacy in the prophylactic treatment of migraine headaches, the following adverse reactions not listed above were reported by 1% or more of patients from two placebo-controlled clinical trials of divalproex sodium tablets.
Body as a Whole: Face edema.
Digestive System: Dry mouth, stomatitis.
Urogenital System: Cystitis, metrorrhagia, and vaginal hemorrhage.
10 Overdosage
Overdosage with valproate may result in somnolence, heart block, deep coma, and hypernatremia. Fatalities have been reported; however patients have recovered from valproate serum concentrations as high as 2,120 mcg/mL.
In overdose situations, the fraction of drug not bound to protein is high and hemodialysis or tandem hemodialysis plus hemoperfusion may result in significant removal of drug. General supportive measures should be applied with particular attention to the maintenance of adequate urinary output.
Naloxone has been reported to reverse the CNS depressant effects of valproate overdosage. Because naloxone could theoretically also reverse the antiepileptic effects of valproate, it should be used with caution in patients with epilepsy.
14.1 Epilepsy
The efficacy of valproate in reducing the incidence of complex partial seizures (CPS) that occur in isolation or in association with other seizure types was established in two controlled trials.
In one, multiclinic, placebo controlled study employing an add-on design (adjunctive therapy), 144 patients who continued to suffer eight or more CPS per 8 weeks during an 8 week period of monotherapy with doses of either carbamazepine or phenytoin sufficient to assure plasma concentrations within the "therapeutic range" were randomized to receive, in addition to their original antiepilepsy drug (AED), either divalproex sodium or placebo. Randomized patients were to be followed for a total of 16 weeks. The following Table presents the findings.
| * Reduction from baseline statistically significantly greater for valproate than placebo at p ≤ 0.05 level. | |||
|
Add-on Treatment |
Number of Patients |
Baseline Incidence |
Experimental Incidence |
|
Divalproex sodium |
75 |
16.0 |
8.9* |
|
Placebo |
69 |
14.5 |
11.5 |
Figure 1 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the adjunctive therapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for an effective treatment is shifted to the left of the curve for placebo. This Figure shows that the proportion of patients achieving any particular level of improvement was consistently higher for valproate than for placebo. For example, 45% of patients treated with valproate had a ≥ 50% reduction in complex partial seizure rate compared to 23% of patients treated with placebo.
The second study assessed the capacity of valproate to reduce the incidence of CPS when administered as the sole AED. The study compared the incidence of CPS among patients randomized to either a high or low dose treatment arm. Patients qualified for entry into the randomized comparison phase of this study only if 1) they continued to experience 2 or more CPS per 4 weeks during an 8 to 12 week long period of monotherapy with adequate doses of an AED (i.e., phenytoin, carbamazepine, phenobarbital, or primidone) and 2) they made a successful transition over a two week interval to valproate. Patients entering the randomized phase were then brought to their assigned target dose, gradually tapered off their concomitant AED and followed for an interval as long as 22 weeks. Less than 50% of the patients randomized, however, completed the study. In patients converted to divalproex sodium monotherapy, the mean total valproate concentrations during monotherapy were 71 and 123 mcg/mL in the low dose and high dose groups, respectively.
The following Table presents the findings for all patients randomized who had at least one post-randomization assessment.
| * Reduction from baseline statistically significantly greater for high dose than low dose at p ≤ 0.05 level. | |||
|
Treatment |
Number of Patients |
Baseline Incidence |
Randomized Phase Incidence |
|
High dose Divalproex sodium |
131 |
13.2 |
10.7* |
|
Low dose Divalproex sodium |
134 |
14.2 |
13.8 |
Figure 2 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the monotherapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for a more effective treatment is shifted to the left of the curve for a less effective treatment. This Figure shows that the proportion of patients achieving any particular level of reduction was consistently higher for high dose valproate than for low dose valproate. For example, when switching from carbamazepine, phenytoin, phenobarbital or primidone monotherapy to high dose valproate monotherapy, 63% of patients experienced no change or a reduction in complex partial seizure rates compared to 54% of patients receiving low dose valproate.
|
Information on pediatric studies is presented in section 8. |
15 References
-
1.Meador KJ, Baker GA, Browning N, et al. Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurology 2013; 12 (3):244-252.
11 Description
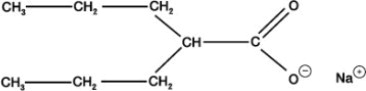
Valproate sodium is the sodium salt of valproic acid designated as sodium 2-propylpentanoate. Valproate sodium has the following structure:
|
C8H15NaO2 |
M.W. 166.2 |
Valproate sodium occurs as an essentially white and odorless, crystalline, deliquescent powder.
Valproate sodium injection, USP is available in 5 mL single-dose vials for intravenous injection. Each mL contains valproate sodium equivalent to 100 mg valproic acid, edetate disodium 0.40 mg, and water for injection to volume. The pH is adjusted to 7.6 with sodium hydroxide and/or hydrochloric acid. The solution is clear and colorless.
7.3 Topiramate
Concomitant administration of valproate and topiramate has been associated with hyperammonemia with and without encephalopathy [see Contraindications (4) and Warnings and Precautions (5.6, 5.8, 5.9)]. Concomitant administration of topiramate with valproate has also been associated with hypothermia in patients who have tolerated either drug alone. It may be prudent to examine blood ammonia levels in patients in whom the onset of hypothermia has been reported [see Warnings and Precautions (5.8, 5.10)].
5.10 Hypothermia
Hypothermia, defined as an unintentional drop in body core temperature to < 35°C (95°F), has been reported in association with valproate therapy both in conjunction with and in the absence of hyperammonemia. This adverse reaction can also occur in patients using concomitant topiramate with valproate after starting topiramate treatment or after increasing the daily dose of topiramate [see Drug Interactions (7.3)]. Consideration should be given to stopping valproate in patients who develop hypothermia, which may be manifested by a variety of clinical abnormalities including lethargy, confusion, coma, and significant alterations in other major organ systems such as the cardiovascular and respiratory systems. Clinical management and assessment should include examination of blood ammonia levels.
5.5 Pancreatitis
Cases of life-threatening pancreatitis have been reported in both children and adults receiving valproate. Some of the cases have been described as hemorrhagic with rapid progression from initial symptoms to death. Some cases have occurred shortly after initial use as well as after several years of use. The rate based upon the reported cases exceeds that expected in the general population and there have been cases in which pancreatitis recurred after rechallenge with valproate. In clinical trials, there were 2 cases of pancreatitis without alternative etiology in 2,416 patients, representing 1,044 patient-years experience. Patients and guardians should be warned that abdominal pain, nausea, vomiting, and/or anorexia can be symptoms of pancreatitis that require prompt medical evaluation. If pancreatitis is diagnosed, valproate should ordinarily be discontinued. Alternative treatment for the underlying medical condition should be initiated as clinically indicated [see Boxed Warning].
8.4 Pediatric Use
Experience with oral valproate has indicated that pediatric patients under the age of two years are at a considerably increased risk of developing fatal hepatotoxicity, especially those with the aforementioned conditions [see Boxed Warning]. The safety of valproate sodium has not been studied in individuals below the age of 2 years. If a decision is made to use valproate sodium in this age group, it should be used with extreme caution and as a sole agent. The benefits of therapy should be weighed against the risks. Above the age of 2 years, experience in epilepsy has indicated that the incidence of fatal hepatotoxicity decreases considerably in progressively older patient groups.
Younger children, especially those receiving enzyme-inducing drugs, will require larger maintenance doses to attain targeted total and unbound valproate concentrations.
The variability in free fraction limits the clinical usefulness of monitoring total serum valproic acid concentrations. Interpretation of valproic acid concentrations in children should include consideration of factors that affect hepatic metabolism and protein binding.
8.5 Geriatric Use
No patients above the age of 65 years were enrolled in double-blind prospective clinical trials of mania associated with bipolar illness. In a case review study of 583 patients, 72 patients (12%) were greater than 65 years of age. A higher percentage of patients above 65 years of age reported accidental injury, infection, pain, somnolence, and tremor. Discontinuation of valproate was occasionally associated with the latter two events. It is not clear whether these events indicate additional risk or whether they result from pre- existing medical illness and concomitant medication use among these patients.
A study of elderly patients with dementia revealed drug related somnolence and discontinuation for somnolence [see Warnings and Precautions (5.13)]. The starting dose should be reduced in these patients, and dosage reductions or discontinuation should be considered in patients with excessive somnolence [see Dosage and Administration (2.2)].
No unique safety concerns were identified in the 21 patients > 65 years of age receiving valproate sodium in clinical trials.
5.8 Hyperammonemia
Hyperammonemia has been reported in association with valproate therapy and may be present despite normal liver function tests. In patients who develop unexplained lethargy and vomiting or changes in mental status, hyperammonemic encephalopathy should be considered and an ammonia level should be measured. Hyperammonemia should also be considered in patients who present with hypothermia [see Warnings and Precautions (5.10)]. If ammonia is increased, valproate therapy should be discontinued. Appropriate interventions for treatment of hyperammonemia should be initiated, and such patients should undergo investigation for underlying urea cycle disorders [see Contraindications (4) and Warnings and Precautions (5.6, 5.9)].
Asymptomatic elevations of ammonia are more common and when present, require close monitoring of plasma ammonia levels. If the elevation persists, discontinuation of valproate therapy should be considered.
14 Clinical Studies
The studies described in the following section were conducted with oral divalproex sodium tablets.
4 Contraindications
-
•Valproate sodium injection should not be administered to patients with hepatic disease or significant hepatic dysfunction [see Warnings and Precautions (5.1)].
-
•Valproate sodium injection is contraindicated in patients known to have mitochondrial disorders caused by mutations in mitochondrial DNA polymerase γ (POLG; e.g., Alpers-Huttenlocher Syndrome) and children under two years of age who are suspected of having a POLG-related disorder [see Warnings and Precautions (5.1)].
-
•Valproate sodium injection is contraindicated in patients with known hypersensitivity to the drug [see Warnings and Precautions (5.11)].
-
•Valproate sodium injection is contraindicated in patients with known urea cycle disorders [see Warnings and Precautions (5.6)].
-
•For use in prophylaxis of migraine headaches: Valproate sodium injection is contraindicated in women who are pregnant and in women of childbearing potential who are not using effective contraception [see Warnings and Precautions (5.2, 5.3, 5.4) and Use in Specific Populations (8.1)].
6 Adverse Reactions
The following serious adverse reactions are described below and elsewhere in the labeling:
-
•Hepatic failure [see Warnings and Precautions (5.1)]
-
•Birth defects [see Warnings and Precautions (5.2)]
-
•Decreased IQ following in utero exposure [see Warnings and Precautions (5.3)]
-
•Pancreatitis [see Warnings and Precautions (5.5)]
-
•Hyperammonemic encephalopathy [see Warnings and Precautions (5.6, 5.8, 5.9)]
-
•Bleeding and other hematopoietic disorders [see Warnings and Precautions (5.7)]
-
•Hypothermia [see Warnings and Precautions (5.10)]
-
•Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan hypersensitivity reactions [see Warnings and Precautions (5.11)]
-
•Somnolence in the elderly [see Warnings and Precautions (5.13)]
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The adverse reactions that can result from valproate sodium use include all of those associated with oral forms of valproate. The following describes experience specifically with valproate sodium. Valproate sodium has been generally well tolerated in clinical trials involving 111 healthy adult male volunteers and 352 patients with epilepsy, given at doses of 125 to 6,000 mg (total daily dose). A total of 2% of patients discontinued treatment with valproate sodium due to adverse reactions. The most common adverse reactions leading to discontinuation were 2 cases each of nausea/vomiting and elevated amylase. Other adverse reactions leading to discontinuation were hallucinations, pneumonia, headache, injection site reaction, and abnormal gait. Dizziness and injection site pain were observed more frequently at a 100 mg/min infusion rate than at rates up to 33 mg/min. At a 200 mg/min rate, dizziness and taste perversion occurred more frequently than at a 100 mg/min rate. The maximum rate of infusion studied was 200 mg/min.
Adverse reactions reported by at least 0.5% of all subjects/patients in clinical trials of valproate sodium are summarized in Table 1.
|
Body System/Reaction |
N = 463
|
|
Body as a Whole |
|
|
Headache |
4.3 |
|
Injection Site Pain |
2.6 |
|
Injection Site Reaction |
2.4 |
|
Chest Pain |
1.7 |
|
Pain (unspecified) |
1.3 |
|
Injection Site Inflammation |
0.6 |
|
Cardiovascular |
|
|
Vasodilation |
0.9 |
|
Dermatologic |
|
|
Sweating |
0.9 |
|
Digestive System |
|
|
Nausea |
3.2 |
|
Vomiting |
1.3 |
|
Abdominal Pain |
1.1 |
|
Diarrhea |
0.9 |
|
Nervous System |
|
|
Dizziness |
5.2 |
|
Somnolence |
1.7 |
|
Euphoria |
0.9 |
|
Nervousness |
0.9 |
|
Paresthesia |
0.9 |
|
Hypesthesia |
0.6 |
|
Tremor |
0.6 |
|
Respiratory |
|
|
Pharyngitis |
0.6 |
|
Special Senses |
|
|
Taste Perversion |
1.9 |
In a separate clinical safety trial, 112 patients with epilepsy were given infusions of valproate (up to 15 mg/kg) over 5 to 10 minutes (1.5 to 3 mg/kg/min). The common adverse reactions (> 2%) were somnolence (10.7%), dizziness (7.1%), paresthesia (7.1%), asthenia (7.1%), nausea (6.3%), and headache (2.7%). While the incidence of these adverse reactions was generally higher than in Table 1 (experience encompassing the standard, much slower infusion rates), e.g., somnolence (1.7%), dizziness (5.2%), paresthesia (0.9%), asthenia (0%), nausea (3.2%), and headache (4.3%), a direct comparison between the incidence of adverse reactions in the 2 cohorts cannot be made because of differences in patient populations and study designs.
Ammonia levels have not been systematically studied after IV valproate, so that an estimate of the incidence of hyperammonemia after IV valproate sodium cannot be provided. Hyperammonemia with encephalopathy has been reported in 2 patients after infusions of valproate sodium.
7 Drug Interactions
-
•Hepatic enzyme-inducing drugs (e.g., phenytoin, carbamazepine, phenobarbital, primidone, rifampin) can increase valproate clearance, while enzyme inhibitors (e.g., felbamate) can decrease valproate clearance. Therefore increased monitoring of valproate and concomitant drug concentrations and dosage adjustment are indicated whenever enzyme-inducing or inhibiting drugs are introduced or withdrawn (7.1)
-
•Aspirin, carbapenem antibiotics, estrogen-containing hormonal contraceptives: Monitoring of valproate concentrations is recommended (7.1)
-
•Co-administration of valproate can affect the pharmacokinetics of other drugs (e.g. diazepam, ethosuximide, lamotrigine, phenytoin) by inhibiting their metabolism or protein binding displacement (7.2)
-
•Patients stabilized on rufinamide should begin valproate therapy at a low dose, and titrate to clinically effective dose (7.2)
-
•Dosage adjustment of amitriptyline/nortriptyline, propofol, warfarin, and zidovudine may be necessary if used concomitantly with valproate (7.2)
-
•Topiramate: Hyperammonemia and encephalopathy (5.9, 7.3)
12.2 Pharmacodynamics
The relationship between plasma concentration and clinical response is not well documented. One contributing factor is the nonlinear, concentration dependent protein binding of valproate which affects the clearance of the drug. Thus, monitoring of total serum valproate cannot provide a reliable index of the bioactive valproate species.
For example, because the plasma protein binding of valproate is concentration dependent, the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Higher than expected free fractions occur in the elderly, in hyperlipidemic patients, and in patients with hepatic and renal diseases.
1 Indications and Usage
Valproate sodium injection is indicated as an intravenous alternative in patients in whom oral administration of valproate products is temporarily not feasible in the following conditions:
-
•Monotherapy and adjunctive therapy of complex partial seizures and simple and complex absence seizures; adjunctive therapy in patients with multiple seizure types that include absence seizures (1)
12.1 Mechanism of Action
Valproate sodium exists as the valproate ion in the blood. The mechanisms by which valproate exerts its therapeutic effects have not been established. It has been suggested that its activity in epilepsy is related to increased brain concentrations of gamma-aminobutyric acid (GABA).
5.6 Urea Cycle Disorders
Valproate sodium is contraindicated in patients with known urea cycle disorders (UCD).
Hyperammonemic encephalopathy, sometimes fatal, has been reported following initiation of valproate therapy in patients with urea cycle disorders, a group of uncommon genetic abnormalities, particularly ornithine transcarbamylase deficiency. Prior to the initiation of valproate therapy, evaluation for UCD should be considered in the following patients: 1) those with a history of unexplained encephalopathy or coma, encephalopathy associated with a protein load, pregnancy-related or postpartum encephalopathy, unexplained mental retardation, or history of elevated plasma ammonia or glutamine; 2) those with cyclical vomiting and lethargy, episodic extreme irritability, ataxia, low BUN, or protein avoidance; 3) those with a family history of UCD or a family history of unexplained infant deaths (particularly males); 4) those with other signs or symptoms of UCD. Patients who develop symptoms of unexplained hyperammonemic encephalopathy while receiving valproate therapy should receive prompt treatment (including discontinuation of valproate therapy) and be evaluated for underlying urea cycle disorders [see Contraindications (4) and Warnings and Precautions (5.9)].
1.2 Important Limitations
Because of the risk to the fetus of decreased IQ, neurodevelopmental disorders, neural tube defects, and other major congenital malformations, which may occur very early in pregnancy, valproate should not be used to treat women with epilepsy or bipolar disorder who are pregnant or who plan to become pregnant unless other medications have failed to provide adequate symptom control or are otherwise unacceptable. Valproate should not be administered to a woman of childbearing potential unless other medications have failed to provide adequate symptom control or are otherwise unacceptable [see Warnings and Precautions (5.2, 5.3, 5.4), Use in Specific Populations (8.1), and Patient Counseling Information (17)].
For prophylaxis of migraine headaches, valproate is contraindicated in women who are pregnant and in women of childbearing potential who are not using effective contraception [see Contraindications (4)].
5 Warnings and Precautions
-
•Hepatotoxicity; evaluate high risk populations and monitor serum liver tests (5.1)
-
•Birth defects, decreased IQ, and neurodevelopmental disorders following in utero exposure; should not be used to treat women with epilepsy or bipolar disorder who are pregnant or who plan to become pregnant or to treat a woman of childbearing potential unless other medications have failed to provide adequate symptom control or are otherwise unacceptable (5.2, 5.3, 5.4)
-
•Pancreatitis; valproate sodium should ordinarily be discontinued (5.5)
-
•Bleeding and other hematopoietic disorders; monitor platelet counts and coagulation tests (5.7)
-
•Hyperammonemia and hyperammonemic encephalopathy; measure ammonia level if unexplained lethargy and vomiting or changes in mental status, and also with concomitant topiramate use; consider discontinuation of valproate therapy (5.6, 5.8, 5.9)
-
•Hypothermia; Hypothermia has been reported during valproate therapy with or without associated hyperammonemia. This adverse reaction can also occur in patients using concomitant topiramate (5.10)
-
•Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan hypersensitivity reaction; discontinue valproate sodium (5.11)
-
•Somnolence in the elderly can occur. Valproate sodium dosage should be increased slowly and with regular monitoring for fluid and nutritional intake (5.13)
2 Dosage and Administration
Valproate sodium injection is intended for intravenous use only.
-
•Epilepsy
-
oComplex Partial Seizures in Adults and Children 10 years of age or older: Initial dose is 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response. Maximum recommended dose is 60 mg/kg/day (2.1).
-
oSimple and Complex Absence Seizures: Initial dose is 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response. Maximum recommended dose is 60 mg/kg/day (2.1).
-
3 Dosage Forms and Strengths
Valproate sodium injection, equivalent to 100 mg of valproic acid per mL, is a clear, colorless solution in 5 mL single-dose vials, available in trays of 10 vials.
Recommended storage: Store vials at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. Preservative Free. Unused portion of container should be discarded.
5.14 Post Traumatic Seizures
A study was conducted to evaluate the effect of IV valproate in the prevention of post-traumatic seizures in patients with acute head injuries. Patients were randomly assigned to receive either IV valproate given for one week (followed by oral valproate products for either one or six months per random treatment assignment) or IV phenytoin given for one week (followed by placebo). In this study, the incidence of death was found to be higher in the two groups assigned to valproate treatment compared to the rate in those assigned to the IV phenytoin treatment group (13% vs. 8.5%, respectively). Many of these patients were critically ill with multiple and/or severe injuries, and evaluation of the causes of death did not suggest any specific drug-related causation. Further, in the absence of a concurrent placebo control during the initial week of intravenous therapy, it is impossible to determine if the mortality rate in the patients treated with valproate was greater or less than that expected in a similar group not treated with valproate, or whether the rate seen in the IV phenytoin treated patients was lower than would be expected. Nonetheless, until further information is available, it seems prudent not to use valproate sodium in patients with acute head trauma for the prophylaxis of post-traumatic seizures.
5.2 Structural Birth Defects
Valproate can cause fetal harm when administered to a pregnant woman. Pregnancy registry data show that maternal valproate use can cause neural tube defects and other structural abnormalities (e.g., craniofacial defects, cardiovascular malformations, hypospadias, limb malformations). The rate of congenital malformations among babies born to mothers using valproate is about four times higher than the rate among babies born to epileptic mothers using other anti-seizure monotherapies. Evidence suggests that folic acid supplementation prior to conception and during the first trimester of pregnancy decreases the risk for congenital neural tube defects in the general population [see Use in Specific Populations (8.1)].
6.4 Postmarketing Experience
The following adverse reactions have been identified during post approval use of divalproex sodium. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatologic: Hair texture changes, hair color changes, photosensitivity, erythema multiforme, toxic epidermal necrolysis, nail and nail bed disorders, and Stevens-Johnson syndrome.
Psychiatric: Emotional upset, psychosis, aggression, psychomotor hyperactivity, hostility, disturbance in attention, learning disorder, and behavioral deterioration.
Neurologic: Paradoxical convulsion, parkinsonism
There have been several reports of acute or subacute cognitive decline and behavioral changes (apathy or irritability) with cerebral pseudoatrophy on imaging associated with valproate therapy; both the cognitive/behavioral changes and cerebral pseudoatrophy reversed partially or fully after valproate discontinuation.
There have been reports of acute or subacute encephalopathy in the absence of elevated ammonia levels, elevated valproate levels, or neuroimaging changes. The encephalopathy reversed partially or fully after valproate discontinuation.
Musculoskeletal: Fractures, decreased bone mineral density, osteopenia, osteoporosis, and weakness.
Hematologic: Relative lymphocytosis, macrocytosis, leucopenia, anemia including macrocytic with or without folate deficiency, bone marrow suppression, pancytopenia, aplastic anemia, agranulocytosis, and acute intermittent porphyria.
Endocrine: Irregular menses, secondary amenorrhea, hyperandrogenism, hirsutism, elevated testosterone level, breast enlargement, galactorrhea, parotid gland swelling, polycystic ovary disease, decreased carnitine concentrations, hyponatremia, hyperglycinemia, and inappropriate ADH secretion.
There have been rare reports of Fanconi's syndrome occurring chiefly in children.
Metabolism and nutrition: Weight gain.
Reproductive: Aspermia, azoospermia, decreased sperm count, decreased spermatozoa motility, male infertility, and abnormal spermatozoa morphology.
Genitourinary: Enuresis and urinary tract infection.
Special Senses: Hearing loss.
Other: Allergic reaction, anaphylaxis, developmental delay, bone pain, bradycardia, and cutaneous vasculitis.
8 Use in Specific Populations
-
•Pregnancy: Valproate sodium can cause congenital malformations including neural tube defects, decreased IQ, and neurodevelopmental disorders (5.2, 5.3, 8.1)
-
•Pediatric: Children under the age of two years are at considerably higher risk of fatal hepatotoxicity (5.1, 8.4)
-
•Geriatric: Reduce starting dose, increase dosage more slowly; monitor fluid and nutritional intake, and somnolence (5.13, 8.5)
5.13 Somnolence in the Elderly
In a double-blind, multicenter trial of valproate in elderly patients with dementia (mean age = 83 years), doses were increased by 125 mg/day to a target dose of 20 mg/kg/day. A significantly higher proportion of valproate patients had somnolence compared to placebo, and although not statistically significant, there was a higher proportion of patients with dehydration. Discontinuations for somnolence were also significantly higher than with placebo. In some patients with somnolence (approximately one-half), there was associated reduced nutritional intake and weight loss. There was a trend for the patients who experienced these events to have a lower baseline albumin concentration, lower valproate clearance, and a higher BUN. In elderly patients, dosage should be increased more slowly and with regular monitoring for fluid and nutritional intake, dehydration, somnolence, and other adverse reactions. Dose reductions or discontinuation of valproate should be considered in patients with decreased food or fluid intake and in patients with excessive somnolence [see Dosage and Administration (2.2)].
5.3 Decreased Iq Following in Utero
Valproate can cause decreased IQ scores following in utero exposure. Published epidemiological studies have indicated that children exposed to valproate in utero have lower cognitive test scores than children exposed in utero to either another antiepileptic drug or to no antiepileptic drugs. The largest of these studies1 is a prospective cohort study conducted in the United States and United Kingdom that found that children with prenatal exposure to valproate (n=62) had lower IQ scores at age 6 (97 [95% C.I. 94 to 101]) than children with prenatal exposure to the other antiepileptic drug monotherapy treatments evaluated: lamotrigine (108 [95% C.I. 105 to 110]), carbamazepine (105 [95% C.I. 102 to 108]), and phenytoin (108 [95% C.I. 104 to 112]). It is not known when during pregnancy cognitive effects in valproate-exposed children occur. Because the women in this study were exposed to antiepileptic drugs throughout pregnancy, whether the risk for decreased IQ was related to a particular time period during pregnancy could not be assessed.
Although all of the available studies have methodological limitations, the weight of the evidence supports the conclusion that valproate exposure in utero can cause decreased IQ in children.
In animal studies, offspring with prenatal exposure to valproate had malformations similar to those seen in humans and demonstrated neurobehavioral deficits [see Use in Specific Populations (8.1)].
16 How Supplied/storage and Handling
Valproate Sodium Injection, equivalent to 100 mg of valproic acid per mL, is a clear, colorless solution supplied as:
|
Manufacturer Product Number |
Unit of Sale |
Strength |
Each Unit |
|
PRX439405 |
NDC 63323-494-16 |
500 mg per 5 mL (100 mg per mL) |
NDC 63323-494-41 |
Store at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature].
Preservative Free. Unused portion of container should be discarded.
The container closure is not made with natural rubber latex.
7.2 Effects of Valproate On Other Drugs
Valproate has been found to be a weak inhibitor of some P450 isozymes, epoxide hydrase, and glucuronosyltransferases.
The following list provides information about the potential for an influence of valproate co-administration on the pharmacokinetics or pharmacodynamics of several commonly prescribed medications. The list is not exhaustive, since new interactions are continuously being reported.
2.3 Dosing in Patients Taking Rufinamide
Patients stabilized on rufinamide before being prescribed valproate should begin valproate therapy at a low dose, and titrate to a clinically effective dose [see Drug Interactions (7.2)].
5.15 Monitoring: Drug Plasma Concentration
Since valproate may interact with concurrently administered drugs which are capable of enzyme induction, periodic plasma concentration determinations of valproate and concomitant drugs are recommended during the early course of therapy [see Drug Interactions (7)].
5.4 Use in Women of Childbearing Potential
Because of the risk to the fetus of decreased IQ, neurodevelopmental disorders, and major congenital malformations (including neural tube defects), which may occur very early in pregnancy, valproate should not be administered to a woman of childbearing potential unless other medications have failed to provide adequate symptom control or are otherwise unacceptable. This is especially important when valproate use is considered for a condition not usually associated with permanent injury or death such as prophylaxis of migraine headaches [see Contraindications (4)]. Women should use effective contraception while using valproate.
Women of childbearing potential should be counseled regularly regarding the relative risks and benefits of valproate use during pregnancy. This is especially important for women planning a pregnancy and for girls at the onset of puberty; alternative therapeutic options should be considered for these patients [see Boxed Warning and Use in Specific Populations (8.1)].
To prevent major seizures, valproate should not be discontinued abruptly, as this can precipitate status epilepticus with resulting maternal and fetal hypoxia and threat to life.
Evidence suggests that folic acid supplementation prior to conception and during the first trimester of pregnancy decreases the risk for congenital neural tube defects in the general population. It is not known whether the risk of neural tube defects or decreased IQ in the offspring of women receiving valproate is reduced by folic acid supplementation. Dietary folic acid supplementation both prior to conception and during pregnancy should be routinely recommended for patients using valproate.
Warning: Life Threatening Adverse Reactions
WARNING: LIFE THREATENING ADVERSE REACTIONS
See full prescribing information for complete boxed warning.
-
•Hepatotoxicity, including fatalities, usually during the first 6 months of treatment. Children under the age of two years and patients with mitochondrial disorders are at higher risk. Monitor patients closely, and perform serum liver testing prior to therapy and at frequent intervals thereafter (5.1)
-
•Fetal Risk, particularly neural tube defects, other major malformations, and decreased IQ (5.2, 5.3, 5.4)
-
•Pancreatitis, including fatal hemorrhagic cases (5.5)
5.12 Interaction With Carbapenem Antibiotics
Carbapenem antibiotics (for example, ertapenem, imipenem, meropenem; this is not a complete list) may reduce serum valproate concentrations to subtherapeutic levels, resulting in loss of seizure control. Serum valproate concentrations should be monitored frequently after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproate concentrations drop significantly or seizure control deteriorates [see Drug Interactions (7.1)].
5.17 Effect On Hiv and Cmv Viruses Replication
There are in vitro studies that suggest valproate stimulates the replication of the HIV and CMV viruses under certain experimental conditions. The clinical consequence, if any, is not known. Additionally, the relevance of these in vitro findings is uncertain for patients receiving maximally suppressive antiretroviral therapy. Nevertheless, these data should be borne in mind when interpreting the results from regular monitoring of the viral load in HIV infected patients receiving valproate or when following CMV infected patients clinically.
5.7 Bleeding and Other Hematopoietic Disorders
Valproate is associated with dose-related thrombocytopenia. In a clinical trial of divalproex sodium as monotherapy in patients with epilepsy, 34/126 patients (27%) receiving approximately 50 mg/kg/day on average, had at least one value of platelets ≤ 75 x 109/L. Approximately half of these patients had treatment discontinued, with return of platelet counts to normal. In the remaining patients, platelet counts normalized with continued treatment. In this study, the probability of thrombocytopenia appeared to increase significantly at total valproate concentrations of ≥ 110 mcg/mL (females) or ≥ 135 mcg/mL (males). The therapeutic benefit which may accompany the higher doses should therefore be weighed against the possibility of a greater incidence of adverse effects. Valproate use has also been associated with decreases in other cell lines and myelodysplasia.
Because of reports of cytopenias, inhibition of the secondary phase of platelet aggregation, and abnormal coagulation parameters (e.g., low fibrinogen, coagulation factor deficiencies, acquired von Willebrand's disease), measurements of complete blood counts and coagulation tests are recommended before initiating therapy and at periodic intervals. It is recommended that patients receiving valproate be monitored for blood counts and coagulation parameters prior to planned surgery and during pregnancy [see Use in Specific Populations (8.1)]. Evidence of hemorrhage, bruising, or a disorder of hemostasis/coagulation is an indication for reduction of the dosage or withdrawal of therapy.
5.16 Effect On Ketone and Thyroid Function Tests
Valproate is partially eliminated in the urine as a keto-metabolite which may lead to a false interpretation of the urine ketone test.
There have been reports of altered thyroid function tests associated with valproate. The clinical significance of these is unknown.
7.1 Effects of Co Administered Drugs On Valproate Clearance
Drugs that affect the level of expression of hepatic enzymes, particularly those that elevate levels of glucuronosyltransferases (such as ritonavir), may increase the clearance of valproate. For example, phenytoin, carbamazepine, and phenobarbital (or primidone) can double the clearance of valproate. Thus, patients on monotherapy will generally have longer half-lives and higher concentrations than patients receiving polytherapy with antiepilepsy drugs.
In contrast, drugs that are inhibitors of cytochrome P450 isozymes, e.g., antidepressants, may be expected to have little effect on valproate clearance because cytochrome P450 microsomal mediated oxidation is a relatively minor secondary metabolic pathway compared to glucuronidation and beta-oxidation.
Because of these changes in valproate clearance, monitoring of valproate and concomitant drug concentrations should be increased whenever enzyme inducing drugs are introduced or withdrawn.
The following list provides information about the potential for an influence of several commonly prescribed medications on valproate pharmacokinetics. The list is not exhaustive nor could it be, since new interactions are continuously being reported.
5.9 Hyperammonemia and Encephalopathy Associated With Concomitant Topiramate Use
Concomitant administration of topiramate and valproate has been associated with hyperammonemia with or without encephalopathy in patients who have tolerated either drug alone. Clinical symptoms of hyperammonemic encephalopathy often include acute alterations in level of consciousness and/or cognitive function with lethargy or vomiting. Hypothermia can also be a manifestation of hyperammonemia [see Warnings and Precautions (5.10)]. In most cases, symptoms and signs abated with discontinuation of either drug. This adverse reaction is not due to a pharmacokinetic interaction. Patients with inborn errors of metabolism or reduced hepatic mitochondrial activity may be at an increased risk for hyperammonemia with or without encephalopathy. Although not studied, an interaction of topiramate and valproate may exacerbate existing defects or unmask deficiencies in susceptible persons. In patients who develop unexplained lethargy, vomiting, or changes in mental status, hyperammonemic encephalopathy should be considered and an ammonia level should be measured [see Contraindications (4) and Warnings and Precautions (5.8)].
Package Label Principal Display Panel Valproate Sodium 5 Ml Single Dose Vial Label
NDC 63323-494-41
Valproate
Sodium
Injection, USP
500 mg* per 5 mL
(100 mg per mL)
For intravenous infusion only.
Preservative free.
5 mL Single-dose Vial Rx only
Package Label Principal Display Panel Valproate Sodium 5 Ml Single Dose Vial Tray Label
NDC 63323-494-16
Valproate
Sodium
Injection, USP
500 mg* per 5 mL
(100 mg per mL)
For intravenous infusion only.
Preservative free.
10 x 5 mL
Single-dose Vials Rx only
5.11 Drug Reaction With Eosinophilia and Systemic Symptoms (dress)/multiorgan Hypersensitivity Reactions
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as Multiorgan Hypersensitivity, has been reported in patients taking valproate. DRESS may be fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy, and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis sometimes resembling an acute viral infection. Eosinophilia is often present. Because this disorder is variable in its expression, other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. Valproate should be discontinued and not be resumed if an alternative etiology for the signs or symptoms cannot be established.
Structured Label Content
Data
Human
Neural tube defects and other structural abnormalities
There is an extensive body of evidence demonstrating that exposure to valproate in utero increases the risk of neural tube defects and other structural abnormalities. Based on published data from the CDC's National Birth Defects Prevention Network, the risk of spina bifida in the general population is about 0.06 to 0.07% (6 to 7 in 10,000 births) compared to the risk following in utero valproate exposure estimated to be approximately 1 to 2% (100 to 200 in 10,000 births).
The NAAED Pregnancy Registry has reported a major malformation rate of 9-11% in the offspring of women exposed to an average of 1,000 mg/day of valproate monotherapy during pregnancy. These data show an up to a five-fold increased risk for any major malformation following valproate exposure in utero compared to the risk following exposure in utero to other AEDs taken as monotherapy. The major congenital malformations included cases of neural tube defects, cardiovascular malformations, craniofacial defects (e.g., oral clefts, craniosynostosis), hypospadias, limb malformations (e.g., clubfoot, polydactyly), and other malformations of varying severity involving other body systems [see Warnings and Precautions (5.2)].
Effect on IQ and neurodevelopmental effects
Published epidemiological studies have indicated that children exposed to valproate in utero have lower IQ scores than children exposed to either another AED in utero or to no AEDs in utero. The largest of these studies1 is a prospective cohort study conducted in the United States and United Kingdom that found that children with prenatal exposure to valproate (n=62) had lower IQ scores at age 6 (97 [95% C.I. 94-101]) than children with prenatal exposure to the other anti-epileptic drug monotherapy treatments evaluated: lamotrigine (108 [95% C.I. 105–110]), carbamazepine (105 [95% C.I. 102–108]) and phenytoin (108 [95% C.I. 104–112]). It is not known when during pregnancy cognitive effects in valproate-exposed children occur. Because the women in this study were exposed to AEDs throughout pregnancy, whether the risk for decreased IQ was related to a particular time period during pregnancy could not be assessed [see Warnings and Precautions (5.3)].
Although the available studies have methodological limitations, the weight of the evidence supports a causal association between valproate exposure in utero and subsequent adverse effects on neurodevelopment, including increases in autism spectrum disorders and attention deficit/hyperactivity disorder (ADHD). An observational study has suggested that exposure to valproate products during pregnancy increases the risk of autism spectrum disorders. In this study, children born to mothers who had used valproate products during pregnancy had 2.9 times the risk (95% confidence interval [CI]: 1.7-4.9) of developing autism spectrum disorders compared to children born to mothers not exposed to valproate products during pregnancy. The absolute risks for autism spectrum disorders were 4.4% (95% CI: 2.6%-7.5%) in valproate-exposed children and 1.5% (95% CI: 1.5%-1.6%) in children not exposed to valproate products. Another observational study found that children who were exposed to valproate in utero had an increased risk of ADHD (adjusted HR 1.48; 95% CI, 1.09-2.00) compared with the unexposed children. Because these studies were observational in nature, conclusions regarding a causal association between in utero valproate exposure and an increased risk of autism spectrum disorder and ADHD cannot be considered definitive.
Other
There are published case reports of fatal hepatic failure in offspring of women who used valproate during pregnancy.
Animal
In developmental toxicity studies conducted in mice, rats, rabbits, and monkeys, increased rates of fetal structural abnormalities, intrauterine growth retardation, and embryo- fetal death occurred following administration of valproate to pregnant animals during organogenesis at clinically relevant doses (calculated on a body surface area [mg/m2] basis). Valproate induced malformations of multiple organ systems, including skeletal, cardiac, and urogenital defects. In mice, in addition to other malformations, fetal neural tube defects have been reported following valproate administration during critical periods of organogenesis, and the teratogenic response correlated with peak maternal drug levels. Behavioral abnormalities (including cognitive, locomotor, and social interaction deficits) and brain histopathological changes have also been reported in mice and rat offspring exposed prenatally to clinically relevant doses of valproate.
Section 42229-5 (42229-5)
Hepatotoxicity
6.2 Mania
Although valproate sodium has not been evaluated for safety and efficacy in the treatment of manic episodes associated with bipolar disorder, the following adverse reactions not listed above were reported by 1% or more of patients from two placebo-controlled clinical trials of divalproex sodium tablets.
Body as a Whole: Chills, neck pain, neck rigidity.
Cardiovascular System: Hypotension, postural hypotension, vasodilation.
Digestive System: Fecal incontinence, gastroenteritis, glossitis.
Musculoskeletal System: Arthrosis.
Nervous System: Agitation, catatonic reaction, hypokinesia, reflexes increased, tardive dyskinesia, vertigo.
Skin and Appendages: Furunculosis, maculopapular rash, seborrhea.
Special Senses: Conjunctivitis, dry eyes, eye pain.
Urogenital: Dysuria.
1.1 Epilepsy
Valproate sodium injection is indicated as an intravenous alternative in patients for whom oral administration of valproate products is temporarily not feasible in the following conditions:
Valproate sodium injection is indicated as monotherapy and adjunctive therapy in the treatment of patients with complex partial seizures that occur either in isolation or in association with other types of seizures. Valproate sodium injection is also indicated for use as sole and adjunctive therapy in the treatment of patients with simple and complex absence seizures, and adjunctively in patients with multiple seizure types that include absence seizures.
Simple absence is defined as very brief clouding of the sensorium or loss of consciousness accompanied by certain generalized epileptic discharges without other detectable clinical signs. Complex absence is the term used when other signs are also present.
See Warnings and Precautions (5.1) for statement regarding fatal hepatic dysfunction.
2.1 Epilepsy
Valproate sodium injection is for intravenous use only.
Use of valproate sodium injection for periods of more than 14 days has not been studied. Patients should be switched to oral valproate products as soon as it is clinically feasible.
Valproate sodium injection should be administered as a 60 minute infusion (but not more than 20 mg/min) with the same frequency as the oral products, although plasma concentration monitoring and dosage adjustments may be necessary.
In one clinical safety study, approximately 90 patients with epilepsy and with no measurable plasma levels of valproate were given single infusions of valproate sodium injection (up to 15 mg/kg and mean dose of 1,184 mg) over 5 to 10 minutes (1.5 to 3 mg/kg/min). Patients generally tolerated the more rapid infusions well [see Adverse Reactions (6.1)]. This study was not designed to assess the effectiveness of these regimens. For pharmacokinetics with rapid infusions, see Clinical Pharmacology (12.3).
6.1 Epilepsy
The data described in the following section were obtained using Depakote (divalproex sodium) tablets.
Based on a placebo-controlled trial of adjunctive therapy for treatment of complex partial seizures, divalproex sodium was generally well tolerated with most adverse reactions rated as mild to moderate in severity. Intolerance was the primary reason for discontinuation in the divalproex sodium-treated patients (6%), compared to 1% of placebo-treated patients.
Table 2 lists treatment-emergent adverse reactions which were reported by ≥ 5% of divalproex sodium-treated patients and for which the incidence was greater than in the placebo group, in the placebo-controlled trial of adjunctive therapy for treatment of complex partial seizures. Since patients were also treated with other antiepilepsy drugs, it is not possible, in most cases, to determine whether the following adverse reactions can be ascribed to divalproex sodium alone, or the combination of divalproex sodium and other antiepilepsy drugs.
|
Body System/Reaction |
Divalproex Sodium %
|
Placebo %
|
|
Body as a Whole |
||
|
Headache |
31 |
21 |
|
Asthenia |
27 |
7 |
|
Fever |
6 |
4 |
|
Gastrointestinal System |
||
|
Nausea |
48 |
14 |
|
Vomiting |
27 |
7 |
|
Abdominal Pain |
23 |
6 |
|
Diarrhea |
13 |
6 |
|
Anorexia |
12 |
0 |
|
Dyspepsia |
8 |
4 |
|
Constipation |
5 |
1 |
|
Nervous System |
||
|
Somnolence |
27 |
11 |
|
Tremor |
25 |
6 |
|
Dizziness |
25 |
13 |
|
Diplopia |
16 |
9 |
|
Amblyopia/Blurred Vision |
12 |
9 |
|
Ataxia |
8 |
1 |
|
Nystagmus |
8 |
1 |
|
Emotional Lability |
6 |
4 |
|
Thinking Abnormal |
6 |
0 |
|
Amnesia |
5 |
1 |
|
Respiratory System |
||
|
Flu Syndrome |
12 |
9 |
|
Infection |
12 |
6 |
|
Bronchitis |
5 |
1 |
|
Rhinitis |
5 |
4 |
|
Other |
||
|
Alopecia |
6 |
1 |
|
Weight Loss |
6 |
0 |
Table 3 lists treatment-emergent adverse reactions which were reported by ≥ 5% of patients in the high dose valproate group, and for which the incidence was greater than in the low dose group, in a controlled trial of divalproex sodium monotherapy treatment of complex partial seizures. Since patients were being titrated off another antiepilepsy drug during the first portion of the trial, it is not possible, in many cases, to determine whether the following adverse reactions can be ascribed to divalproex sodium alone, or the combination of valproate and other antiepilepsy drugs.
|
Body System/Reaction |
High Dose %
|
Low Dose %
|
|
Body as a Whole |
||
|
Asthenia |
21 |
10 |
|
Nausea |
34 |
26 |
|
Diarrhea |
23 |
19 |
|
Vomiting |
23 |
15 |
|
Abdominal Pain |
12 |
9 |
|
Anorexia |
11 |
4 |
|
Dyspepsia |
11 |
10 |
|
Thrombocytopenia |
24 |
1 |
|
Ecchymosis |
5 |
4 |
|
Weight Gain |
9 |
4 |
|
Peripheral Edema |
8 |
3 |
|
Tremor |
57 |
19 |
|
Somnolence |
30 |
18 |
|
Dizziness |
18 |
13 |
|
Insomnia |
15 |
9 |
|
Nervousness |
11 |
7 |
|
Amnesia |
7 |
4 |
|
Nystagmus |
7 |
1 |
|
Depression |
5 |
4 |
|
Infection |
20 |
13 |
|
Pharyngitis |
8 |
2 |
|
Dyspnea |
5 |
1 |
|
Alopecia |
24 |
13 |
|
Special Senses |
||
|
Amblyopia/Blurred Vision |
8 |
4 |
|
Tinnitus |
7 |
1 |
1 Headache was the only adverse reaction that occurred in ≥ 5% of patients in the high dose group and at an equal or greater incidence in the low dose group.
The following additional adverse reactions were reported by greater than 1% but less than 5% of the 358 patients treated with valproate in the controlled trials of complex partial seizures:
Body as a Whole: Back pain, chest pain, malaise.
Cardiovascular System: Tachycardia, hypertension, palpitation.
Digestive System: Increased appetite, flatulence, hematemesis, eructation, pancreatitis, periodontal abscess.
Hemic and Lymphatic System: Petechia.
Metabolic and Nutritional Disorders: SGOT increased, SGPT increased.
Musculoskeletal System: Myalgia, twitching, arthralgia, leg cramps, myasthenia.
Nervous System: Anxiety, confusion, abnormal gait, paresthesia, hypertonia, incoordination, abnormal dreams, personality disorder.
Respiratory System: Sinusitis, cough increased, pneumonia, epistaxis.
Skin and Appendages: Rash, pruritus, dry skin.
Special Senses: Taste perversion, abnormal vision, deafness, otitis media.
Urogenital System: Urinary incontinence, vaginitis, dysmenorrhea, amenorrhea, urinary frequency.
6.3 Migraine
Although valproate has not been evaluated for safety and efficacy in the prophylactic treatment of migraine headaches, the following adverse reactions not listed above were reported by 1% or more of patients from two placebo-controlled clinical trials of divalproex sodium tablets.
Body as a Whole: Face edema.
Digestive System: Dry mouth, stomatitis.
Urogenital System: Cystitis, metrorrhagia, and vaginal hemorrhage.
10 Overdosage (10 OVERDOSAGE)
Overdosage with valproate may result in somnolence, heart block, deep coma, and hypernatremia. Fatalities have been reported; however patients have recovered from valproate serum concentrations as high as 2,120 mcg/mL.
In overdose situations, the fraction of drug not bound to protein is high and hemodialysis or tandem hemodialysis plus hemoperfusion may result in significant removal of drug. General supportive measures should be applied with particular attention to the maintenance of adequate urinary output.
Naloxone has been reported to reverse the CNS depressant effects of valproate overdosage. Because naloxone could theoretically also reverse the antiepileptic effects of valproate, it should be used with caution in patients with epilepsy.
14.1 Epilepsy
The efficacy of valproate in reducing the incidence of complex partial seizures (CPS) that occur in isolation or in association with other seizure types was established in two controlled trials.
In one, multiclinic, placebo controlled study employing an add-on design (adjunctive therapy), 144 patients who continued to suffer eight or more CPS per 8 weeks during an 8 week period of monotherapy with doses of either carbamazepine or phenytoin sufficient to assure plasma concentrations within the "therapeutic range" were randomized to receive, in addition to their original antiepilepsy drug (AED), either divalproex sodium or placebo. Randomized patients were to be followed for a total of 16 weeks. The following Table presents the findings.
| * Reduction from baseline statistically significantly greater for valproate than placebo at p ≤ 0.05 level. | |||
|
Add-on Treatment |
Number of Patients |
Baseline Incidence |
Experimental Incidence |
|
Divalproex sodium |
75 |
16.0 |
8.9* |
|
Placebo |
69 |
14.5 |
11.5 |
Figure 1 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the adjunctive therapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for an effective treatment is shifted to the left of the curve for placebo. This Figure shows that the proportion of patients achieving any particular level of improvement was consistently higher for valproate than for placebo. For example, 45% of patients treated with valproate had a ≥ 50% reduction in complex partial seizure rate compared to 23% of patients treated with placebo.
The second study assessed the capacity of valproate to reduce the incidence of CPS when administered as the sole AED. The study compared the incidence of CPS among patients randomized to either a high or low dose treatment arm. Patients qualified for entry into the randomized comparison phase of this study only if 1) they continued to experience 2 or more CPS per 4 weeks during an 8 to 12 week long period of monotherapy with adequate doses of an AED (i.e., phenytoin, carbamazepine, phenobarbital, or primidone) and 2) they made a successful transition over a two week interval to valproate. Patients entering the randomized phase were then brought to their assigned target dose, gradually tapered off their concomitant AED and followed for an interval as long as 22 weeks. Less than 50% of the patients randomized, however, completed the study. In patients converted to divalproex sodium monotherapy, the mean total valproate concentrations during monotherapy were 71 and 123 mcg/mL in the low dose and high dose groups, respectively.
The following Table presents the findings for all patients randomized who had at least one post-randomization assessment.
| * Reduction from baseline statistically significantly greater for high dose than low dose at p ≤ 0.05 level. | |||
|
Treatment |
Number of Patients |
Baseline Incidence |
Randomized Phase Incidence |
|
High dose Divalproex sodium |
131 |
13.2 |
10.7* |
|
Low dose Divalproex sodium |
134 |
14.2 |
13.8 |
Figure 2 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the monotherapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for a more effective treatment is shifted to the left of the curve for a less effective treatment. This Figure shows that the proportion of patients achieving any particular level of reduction was consistently higher for high dose valproate than for low dose valproate. For example, when switching from carbamazepine, phenytoin, phenobarbital or primidone monotherapy to high dose valproate monotherapy, 63% of patients experienced no change or a reduction in complex partial seizure rates compared to 54% of patients receiving low dose valproate.
|
Information on pediatric studies is presented in section 8. |
15 References (15 REFERENCES)
-
1.Meador KJ, Baker GA, Browning N, et al. Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurology 2013; 12 (3):244-252.
11 Description (11 DESCRIPTION)
Valproate sodium is the sodium salt of valproic acid designated as sodium 2-propylpentanoate. Valproate sodium has the following structure:
|
C8H15NaO2 |
M.W. 166.2 |
Valproate sodium occurs as an essentially white and odorless, crystalline, deliquescent powder.
Valproate sodium injection, USP is available in 5 mL single-dose vials for intravenous injection. Each mL contains valproate sodium equivalent to 100 mg valproic acid, edetate disodium 0.40 mg, and water for injection to volume. The pH is adjusted to 7.6 with sodium hydroxide and/or hydrochloric acid. The solution is clear and colorless.
7.3 Topiramate
Concomitant administration of valproate and topiramate has been associated with hyperammonemia with and without encephalopathy [see Contraindications (4) and Warnings and Precautions (5.6, 5.8, 5.9)]. Concomitant administration of topiramate with valproate has also been associated with hypothermia in patients who have tolerated either drug alone. It may be prudent to examine blood ammonia levels in patients in whom the onset of hypothermia has been reported [see Warnings and Precautions (5.8, 5.10)].
5.10 Hypothermia
Hypothermia, defined as an unintentional drop in body core temperature to < 35°C (95°F), has been reported in association with valproate therapy both in conjunction with and in the absence of hyperammonemia. This adverse reaction can also occur in patients using concomitant topiramate with valproate after starting topiramate treatment or after increasing the daily dose of topiramate [see Drug Interactions (7.3)]. Consideration should be given to stopping valproate in patients who develop hypothermia, which may be manifested by a variety of clinical abnormalities including lethargy, confusion, coma, and significant alterations in other major organ systems such as the cardiovascular and respiratory systems. Clinical management and assessment should include examination of blood ammonia levels.
5.5 Pancreatitis
Cases of life-threatening pancreatitis have been reported in both children and adults receiving valproate. Some of the cases have been described as hemorrhagic with rapid progression from initial symptoms to death. Some cases have occurred shortly after initial use as well as after several years of use. The rate based upon the reported cases exceeds that expected in the general population and there have been cases in which pancreatitis recurred after rechallenge with valproate. In clinical trials, there were 2 cases of pancreatitis without alternative etiology in 2,416 patients, representing 1,044 patient-years experience. Patients and guardians should be warned that abdominal pain, nausea, vomiting, and/or anorexia can be symptoms of pancreatitis that require prompt medical evaluation. If pancreatitis is diagnosed, valproate should ordinarily be discontinued. Alternative treatment for the underlying medical condition should be initiated as clinically indicated [see Boxed Warning].
8.4 Pediatric Use
Experience with oral valproate has indicated that pediatric patients under the age of two years are at a considerably increased risk of developing fatal hepatotoxicity, especially those with the aforementioned conditions [see Boxed Warning]. The safety of valproate sodium has not been studied in individuals below the age of 2 years. If a decision is made to use valproate sodium in this age group, it should be used with extreme caution and as a sole agent. The benefits of therapy should be weighed against the risks. Above the age of 2 years, experience in epilepsy has indicated that the incidence of fatal hepatotoxicity decreases considerably in progressively older patient groups.
Younger children, especially those receiving enzyme-inducing drugs, will require larger maintenance doses to attain targeted total and unbound valproate concentrations.
The variability in free fraction limits the clinical usefulness of monitoring total serum valproic acid concentrations. Interpretation of valproic acid concentrations in children should include consideration of factors that affect hepatic metabolism and protein binding.
8.5 Geriatric Use
No patients above the age of 65 years were enrolled in double-blind prospective clinical trials of mania associated with bipolar illness. In a case review study of 583 patients, 72 patients (12%) were greater than 65 years of age. A higher percentage of patients above 65 years of age reported accidental injury, infection, pain, somnolence, and tremor. Discontinuation of valproate was occasionally associated with the latter two events. It is not clear whether these events indicate additional risk or whether they result from pre- existing medical illness and concomitant medication use among these patients.
A study of elderly patients with dementia revealed drug related somnolence and discontinuation for somnolence [see Warnings and Precautions (5.13)]. The starting dose should be reduced in these patients, and dosage reductions or discontinuation should be considered in patients with excessive somnolence [see Dosage and Administration (2.2)].
No unique safety concerns were identified in the 21 patients > 65 years of age receiving valproate sodium in clinical trials.
5.8 Hyperammonemia
Hyperammonemia has been reported in association with valproate therapy and may be present despite normal liver function tests. In patients who develop unexplained lethargy and vomiting or changes in mental status, hyperammonemic encephalopathy should be considered and an ammonia level should be measured. Hyperammonemia should also be considered in patients who present with hypothermia [see Warnings and Precautions (5.10)]. If ammonia is increased, valproate therapy should be discontinued. Appropriate interventions for treatment of hyperammonemia should be initiated, and such patients should undergo investigation for underlying urea cycle disorders [see Contraindications (4) and Warnings and Precautions (5.6, 5.9)].
Asymptomatic elevations of ammonia are more common and when present, require close monitoring of plasma ammonia levels. If the elevation persists, discontinuation of valproate therapy should be considered.
14 Clinical Studies (14 CLINICAL STUDIES)
The studies described in the following section were conducted with oral divalproex sodium tablets.
4 Contraindications (4 CONTRAINDICATIONS)
-
•Valproate sodium injection should not be administered to patients with hepatic disease or significant hepatic dysfunction [see Warnings and Precautions (5.1)].
-
•Valproate sodium injection is contraindicated in patients known to have mitochondrial disorders caused by mutations in mitochondrial DNA polymerase γ (POLG; e.g., Alpers-Huttenlocher Syndrome) and children under two years of age who are suspected of having a POLG-related disorder [see Warnings and Precautions (5.1)].
-
•Valproate sodium injection is contraindicated in patients with known hypersensitivity to the drug [see Warnings and Precautions (5.11)].
-
•Valproate sodium injection is contraindicated in patients with known urea cycle disorders [see Warnings and Precautions (5.6)].
-
•For use in prophylaxis of migraine headaches: Valproate sodium injection is contraindicated in women who are pregnant and in women of childbearing potential who are not using effective contraception [see Warnings and Precautions (5.2, 5.3, 5.4) and Use in Specific Populations (8.1)].
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following serious adverse reactions are described below and elsewhere in the labeling:
-
•Hepatic failure [see Warnings and Precautions (5.1)]
-
•Birth defects [see Warnings and Precautions (5.2)]
-
•Decreased IQ following in utero exposure [see Warnings and Precautions (5.3)]
-
•Pancreatitis [see Warnings and Precautions (5.5)]
-
•Hyperammonemic encephalopathy [see Warnings and Precautions (5.6, 5.8, 5.9)]
-
•Bleeding and other hematopoietic disorders [see Warnings and Precautions (5.7)]
-
•Hypothermia [see Warnings and Precautions (5.10)]
-
•Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan hypersensitivity reactions [see Warnings and Precautions (5.11)]
-
•Somnolence in the elderly [see Warnings and Precautions (5.13)]
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The adverse reactions that can result from valproate sodium use include all of those associated with oral forms of valproate. The following describes experience specifically with valproate sodium. Valproate sodium has been generally well tolerated in clinical trials involving 111 healthy adult male volunteers and 352 patients with epilepsy, given at doses of 125 to 6,000 mg (total daily dose). A total of 2% of patients discontinued treatment with valproate sodium due to adverse reactions. The most common adverse reactions leading to discontinuation were 2 cases each of nausea/vomiting and elevated amylase. Other adverse reactions leading to discontinuation were hallucinations, pneumonia, headache, injection site reaction, and abnormal gait. Dizziness and injection site pain were observed more frequently at a 100 mg/min infusion rate than at rates up to 33 mg/min. At a 200 mg/min rate, dizziness and taste perversion occurred more frequently than at a 100 mg/min rate. The maximum rate of infusion studied was 200 mg/min.
Adverse reactions reported by at least 0.5% of all subjects/patients in clinical trials of valproate sodium are summarized in Table 1.
|
Body System/Reaction |
N = 463
|
|
Body as a Whole |
|
|
Headache |
4.3 |
|
Injection Site Pain |
2.6 |
|
Injection Site Reaction |
2.4 |
|
Chest Pain |
1.7 |
|
Pain (unspecified) |
1.3 |
|
Injection Site Inflammation |
0.6 |
|
Cardiovascular |
|
|
Vasodilation |
0.9 |
|
Dermatologic |
|
|
Sweating |
0.9 |
|
Digestive System |
|
|
Nausea |
3.2 |
|
Vomiting |
1.3 |
|
Abdominal Pain |
1.1 |
|
Diarrhea |
0.9 |
|
Nervous System |
|
|
Dizziness |
5.2 |
|
Somnolence |
1.7 |
|
Euphoria |
0.9 |
|
Nervousness |
0.9 |
|
Paresthesia |
0.9 |
|
Hypesthesia |
0.6 |
|
Tremor |
0.6 |
|
Respiratory |
|
|
Pharyngitis |
0.6 |
|
Special Senses |
|
|
Taste Perversion |
1.9 |
In a separate clinical safety trial, 112 patients with epilepsy were given infusions of valproate (up to 15 mg/kg) over 5 to 10 minutes (1.5 to 3 mg/kg/min). The common adverse reactions (> 2%) were somnolence (10.7%), dizziness (7.1%), paresthesia (7.1%), asthenia (7.1%), nausea (6.3%), and headache (2.7%). While the incidence of these adverse reactions was generally higher than in Table 1 (experience encompassing the standard, much slower infusion rates), e.g., somnolence (1.7%), dizziness (5.2%), paresthesia (0.9%), asthenia (0%), nausea (3.2%), and headache (4.3%), a direct comparison between the incidence of adverse reactions in the 2 cohorts cannot be made because of differences in patient populations and study designs.
Ammonia levels have not been systematically studied after IV valproate, so that an estimate of the incidence of hyperammonemia after IV valproate sodium cannot be provided. Hyperammonemia with encephalopathy has been reported in 2 patients after infusions of valproate sodium.
7 Drug Interactions (7 DRUG INTERACTIONS)
-
•Hepatic enzyme-inducing drugs (e.g., phenytoin, carbamazepine, phenobarbital, primidone, rifampin) can increase valproate clearance, while enzyme inhibitors (e.g., felbamate) can decrease valproate clearance. Therefore increased monitoring of valproate and concomitant drug concentrations and dosage adjustment are indicated whenever enzyme-inducing or inhibiting drugs are introduced or withdrawn (7.1)
-
•Aspirin, carbapenem antibiotics, estrogen-containing hormonal contraceptives: Monitoring of valproate concentrations is recommended (7.1)
-
•Co-administration of valproate can affect the pharmacokinetics of other drugs (e.g. diazepam, ethosuximide, lamotrigine, phenytoin) by inhibiting their metabolism or protein binding displacement (7.2)
-
•Patients stabilized on rufinamide should begin valproate therapy at a low dose, and titrate to clinically effective dose (7.2)
-
•Dosage adjustment of amitriptyline/nortriptyline, propofol, warfarin, and zidovudine may be necessary if used concomitantly with valproate (7.2)
-
•Topiramate: Hyperammonemia and encephalopathy (5.9, 7.3)
12.2 Pharmacodynamics
The relationship between plasma concentration and clinical response is not well documented. One contributing factor is the nonlinear, concentration dependent protein binding of valproate which affects the clearance of the drug. Thus, monitoring of total serum valproate cannot provide a reliable index of the bioactive valproate species.
For example, because the plasma protein binding of valproate is concentration dependent, the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Higher than expected free fractions occur in the elderly, in hyperlipidemic patients, and in patients with hepatic and renal diseases.
1 Indications and Usage (1 INDICATIONS AND USAGE)
Valproate sodium injection is indicated as an intravenous alternative in patients in whom oral administration of valproate products is temporarily not feasible in the following conditions:
-
•Monotherapy and adjunctive therapy of complex partial seizures and simple and complex absence seizures; adjunctive therapy in patients with multiple seizure types that include absence seizures (1)
12.1 Mechanism of Action
Valproate sodium exists as the valproate ion in the blood. The mechanisms by which valproate exerts its therapeutic effects have not been established. It has been suggested that its activity in epilepsy is related to increased brain concentrations of gamma-aminobutyric acid (GABA).
5.6 Urea Cycle Disorders
Valproate sodium is contraindicated in patients with known urea cycle disorders (UCD).
Hyperammonemic encephalopathy, sometimes fatal, has been reported following initiation of valproate therapy in patients with urea cycle disorders, a group of uncommon genetic abnormalities, particularly ornithine transcarbamylase deficiency. Prior to the initiation of valproate therapy, evaluation for UCD should be considered in the following patients: 1) those with a history of unexplained encephalopathy or coma, encephalopathy associated with a protein load, pregnancy-related or postpartum encephalopathy, unexplained mental retardation, or history of elevated plasma ammonia or glutamine; 2) those with cyclical vomiting and lethargy, episodic extreme irritability, ataxia, low BUN, or protein avoidance; 3) those with a family history of UCD or a family history of unexplained infant deaths (particularly males); 4) those with other signs or symptoms of UCD. Patients who develop symptoms of unexplained hyperammonemic encephalopathy while receiving valproate therapy should receive prompt treatment (including discontinuation of valproate therapy) and be evaluated for underlying urea cycle disorders [see Contraindications (4) and Warnings and Precautions (5.9)].
1.2 Important Limitations
Because of the risk to the fetus of decreased IQ, neurodevelopmental disorders, neural tube defects, and other major congenital malformations, which may occur very early in pregnancy, valproate should not be used to treat women with epilepsy or bipolar disorder who are pregnant or who plan to become pregnant unless other medications have failed to provide adequate symptom control or are otherwise unacceptable. Valproate should not be administered to a woman of childbearing potential unless other medications have failed to provide adequate symptom control or are otherwise unacceptable [see Warnings and Precautions (5.2, 5.3, 5.4), Use in Specific Populations (8.1), and Patient Counseling Information (17)].
For prophylaxis of migraine headaches, valproate is contraindicated in women who are pregnant and in women of childbearing potential who are not using effective contraception [see Contraindications (4)].
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
-
•Hepatotoxicity; evaluate high risk populations and monitor serum liver tests (5.1)
-
•Birth defects, decreased IQ, and neurodevelopmental disorders following in utero exposure; should not be used to treat women with epilepsy or bipolar disorder who are pregnant or who plan to become pregnant or to treat a woman of childbearing potential unless other medications have failed to provide adequate symptom control or are otherwise unacceptable (5.2, 5.3, 5.4)
-
•Pancreatitis; valproate sodium should ordinarily be discontinued (5.5)
-
•Bleeding and other hematopoietic disorders; monitor platelet counts and coagulation tests (5.7)
-
•Hyperammonemia and hyperammonemic encephalopathy; measure ammonia level if unexplained lethargy and vomiting or changes in mental status, and also with concomitant topiramate use; consider discontinuation of valproate therapy (5.6, 5.8, 5.9)
-
•Hypothermia; Hypothermia has been reported during valproate therapy with or without associated hyperammonemia. This adverse reaction can also occur in patients using concomitant topiramate (5.10)
-
•Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan hypersensitivity reaction; discontinue valproate sodium (5.11)
-
•Somnolence in the elderly can occur. Valproate sodium dosage should be increased slowly and with regular monitoring for fluid and nutritional intake (5.13)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
Valproate sodium injection is intended for intravenous use only.
-
•Epilepsy
-
oComplex Partial Seizures in Adults and Children 10 years of age or older: Initial dose is 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response. Maximum recommended dose is 60 mg/kg/day (2.1).
-
oSimple and Complex Absence Seizures: Initial dose is 10 to 15 mg/kg/day, increasing at 1 week intervals by 5 to 10 mg/kg/day to achieve optimal clinical response. Maximum recommended dose is 60 mg/kg/day (2.1).
-
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Valproate sodium injection, equivalent to 100 mg of valproic acid per mL, is a clear, colorless solution in 5 mL single-dose vials, available in trays of 10 vials.
Recommended storage: Store vials at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature]. Preservative Free. Unused portion of container should be discarded.
5.14 Post Traumatic Seizures (5.14 Post-traumatic Seizures)
A study was conducted to evaluate the effect of IV valproate in the prevention of post-traumatic seizures in patients with acute head injuries. Patients were randomly assigned to receive either IV valproate given for one week (followed by oral valproate products for either one or six months per random treatment assignment) or IV phenytoin given for one week (followed by placebo). In this study, the incidence of death was found to be higher in the two groups assigned to valproate treatment compared to the rate in those assigned to the IV phenytoin treatment group (13% vs. 8.5%, respectively). Many of these patients were critically ill with multiple and/or severe injuries, and evaluation of the causes of death did not suggest any specific drug-related causation. Further, in the absence of a concurrent placebo control during the initial week of intravenous therapy, it is impossible to determine if the mortality rate in the patients treated with valproate was greater or less than that expected in a similar group not treated with valproate, or whether the rate seen in the IV phenytoin treated patients was lower than would be expected. Nonetheless, until further information is available, it seems prudent not to use valproate sodium in patients with acute head trauma for the prophylaxis of post-traumatic seizures.
5.2 Structural Birth Defects
Valproate can cause fetal harm when administered to a pregnant woman. Pregnancy registry data show that maternal valproate use can cause neural tube defects and other structural abnormalities (e.g., craniofacial defects, cardiovascular malformations, hypospadias, limb malformations). The rate of congenital malformations among babies born to mothers using valproate is about four times higher than the rate among babies born to epileptic mothers using other anti-seizure monotherapies. Evidence suggests that folic acid supplementation prior to conception and during the first trimester of pregnancy decreases the risk for congenital neural tube defects in the general population [see Use in Specific Populations (8.1)].
6.4 Postmarketing Experience
The following adverse reactions have been identified during post approval use of divalproex sodium. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatologic: Hair texture changes, hair color changes, photosensitivity, erythema multiforme, toxic epidermal necrolysis, nail and nail bed disorders, and Stevens-Johnson syndrome.
Psychiatric: Emotional upset, psychosis, aggression, psychomotor hyperactivity, hostility, disturbance in attention, learning disorder, and behavioral deterioration.
Neurologic: Paradoxical convulsion, parkinsonism
There have been several reports of acute or subacute cognitive decline and behavioral changes (apathy or irritability) with cerebral pseudoatrophy on imaging associated with valproate therapy; both the cognitive/behavioral changes and cerebral pseudoatrophy reversed partially or fully after valproate discontinuation.
There have been reports of acute or subacute encephalopathy in the absence of elevated ammonia levels, elevated valproate levels, or neuroimaging changes. The encephalopathy reversed partially or fully after valproate discontinuation.
Musculoskeletal: Fractures, decreased bone mineral density, osteopenia, osteoporosis, and weakness.
Hematologic: Relative lymphocytosis, macrocytosis, leucopenia, anemia including macrocytic with or without folate deficiency, bone marrow suppression, pancytopenia, aplastic anemia, agranulocytosis, and acute intermittent porphyria.
Endocrine: Irregular menses, secondary amenorrhea, hyperandrogenism, hirsutism, elevated testosterone level, breast enlargement, galactorrhea, parotid gland swelling, polycystic ovary disease, decreased carnitine concentrations, hyponatremia, hyperglycinemia, and inappropriate ADH secretion.
There have been rare reports of Fanconi's syndrome occurring chiefly in children.
Metabolism and nutrition: Weight gain.
Reproductive: Aspermia, azoospermia, decreased sperm count, decreased spermatozoa motility, male infertility, and abnormal spermatozoa morphology.
Genitourinary: Enuresis and urinary tract infection.
Special Senses: Hearing loss.
Other: Allergic reaction, anaphylaxis, developmental delay, bone pain, bradycardia, and cutaneous vasculitis.
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
-
•Pregnancy: Valproate sodium can cause congenital malformations including neural tube defects, decreased IQ, and neurodevelopmental disorders (5.2, 5.3, 8.1)
-
•Pediatric: Children under the age of two years are at considerably higher risk of fatal hepatotoxicity (5.1, 8.4)
-
•Geriatric: Reduce starting dose, increase dosage more slowly; monitor fluid and nutritional intake, and somnolence (5.13, 8.5)
5.13 Somnolence in the Elderly
In a double-blind, multicenter trial of valproate in elderly patients with dementia (mean age = 83 years), doses were increased by 125 mg/day to a target dose of 20 mg/kg/day. A significantly higher proportion of valproate patients had somnolence compared to placebo, and although not statistically significant, there was a higher proportion of patients with dehydration. Discontinuations for somnolence were also significantly higher than with placebo. In some patients with somnolence (approximately one-half), there was associated reduced nutritional intake and weight loss. There was a trend for the patients who experienced these events to have a lower baseline albumin concentration, lower valproate clearance, and a higher BUN. In elderly patients, dosage should be increased more slowly and with regular monitoring for fluid and nutritional intake, dehydration, somnolence, and other adverse reactions. Dose reductions or discontinuation of valproate should be considered in patients with decreased food or fluid intake and in patients with excessive somnolence [see Dosage and Administration (2.2)].
5.3 Decreased Iq Following in Utero (5.3 Decreased IQ Following in utero)
Valproate can cause decreased IQ scores following in utero exposure. Published epidemiological studies have indicated that children exposed to valproate in utero have lower cognitive test scores than children exposed in utero to either another antiepileptic drug or to no antiepileptic drugs. The largest of these studies1 is a prospective cohort study conducted in the United States and United Kingdom that found that children with prenatal exposure to valproate (n=62) had lower IQ scores at age 6 (97 [95% C.I. 94 to 101]) than children with prenatal exposure to the other antiepileptic drug monotherapy treatments evaluated: lamotrigine (108 [95% C.I. 105 to 110]), carbamazepine (105 [95% C.I. 102 to 108]), and phenytoin (108 [95% C.I. 104 to 112]). It is not known when during pregnancy cognitive effects in valproate-exposed children occur. Because the women in this study were exposed to antiepileptic drugs throughout pregnancy, whether the risk for decreased IQ was related to a particular time period during pregnancy could not be assessed.
Although all of the available studies have methodological limitations, the weight of the evidence supports the conclusion that valproate exposure in utero can cause decreased IQ in children.
In animal studies, offspring with prenatal exposure to valproate had malformations similar to those seen in humans and demonstrated neurobehavioral deficits [see Use in Specific Populations (8.1)].
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
Valproate Sodium Injection, equivalent to 100 mg of valproic acid per mL, is a clear, colorless solution supplied as:
|
Manufacturer Product Number |
Unit of Sale |
Strength |
Each Unit |
|
PRX439405 |
NDC 63323-494-16 |
500 mg per 5 mL (100 mg per mL) |
NDC 63323-494-41 |
Store at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature].
Preservative Free. Unused portion of container should be discarded.
The container closure is not made with natural rubber latex.
7.2 Effects of Valproate On Other Drugs (7.2 Effects of Valproate on Other Drugs)
Valproate has been found to be a weak inhibitor of some P450 isozymes, epoxide hydrase, and glucuronosyltransferases.
The following list provides information about the potential for an influence of valproate co-administration on the pharmacokinetics or pharmacodynamics of several commonly prescribed medications. The list is not exhaustive, since new interactions are continuously being reported.
2.3 Dosing in Patients Taking Rufinamide
Patients stabilized on rufinamide before being prescribed valproate should begin valproate therapy at a low dose, and titrate to a clinically effective dose [see Drug Interactions (7.2)].
5.15 Monitoring: Drug Plasma Concentration
Since valproate may interact with concurrently administered drugs which are capable of enzyme induction, periodic plasma concentration determinations of valproate and concomitant drugs are recommended during the early course of therapy [see Drug Interactions (7)].
5.4 Use in Women of Childbearing Potential
Because of the risk to the fetus of decreased IQ, neurodevelopmental disorders, and major congenital malformations (including neural tube defects), which may occur very early in pregnancy, valproate should not be administered to a woman of childbearing potential unless other medications have failed to provide adequate symptom control or are otherwise unacceptable. This is especially important when valproate use is considered for a condition not usually associated with permanent injury or death such as prophylaxis of migraine headaches [see Contraindications (4)]. Women should use effective contraception while using valproate.
Women of childbearing potential should be counseled regularly regarding the relative risks and benefits of valproate use during pregnancy. This is especially important for women planning a pregnancy and for girls at the onset of puberty; alternative therapeutic options should be considered for these patients [see Boxed Warning and Use in Specific Populations (8.1)].
To prevent major seizures, valproate should not be discontinued abruptly, as this can precipitate status epilepticus with resulting maternal and fetal hypoxia and threat to life.
Evidence suggests that folic acid supplementation prior to conception and during the first trimester of pregnancy decreases the risk for congenital neural tube defects in the general population. It is not known whether the risk of neural tube defects or decreased IQ in the offspring of women receiving valproate is reduced by folic acid supplementation. Dietary folic acid supplementation both prior to conception and during pregnancy should be routinely recommended for patients using valproate.
Warning: Life Threatening Adverse Reactions (WARNING: LIFE THREATENING ADVERSE REACTIONS)
WARNING: LIFE THREATENING ADVERSE REACTIONS
See full prescribing information for complete boxed warning.
-
•Hepatotoxicity, including fatalities, usually during the first 6 months of treatment. Children under the age of two years and patients with mitochondrial disorders are at higher risk. Monitor patients closely, and perform serum liver testing prior to therapy and at frequent intervals thereafter (5.1)
-
•Fetal Risk, particularly neural tube defects, other major malformations, and decreased IQ (5.2, 5.3, 5.4)
-
•Pancreatitis, including fatal hemorrhagic cases (5.5)
5.12 Interaction With Carbapenem Antibiotics (5.12 Interaction with Carbapenem Antibiotics)
Carbapenem antibiotics (for example, ertapenem, imipenem, meropenem; this is not a complete list) may reduce serum valproate concentrations to subtherapeutic levels, resulting in loss of seizure control. Serum valproate concentrations should be monitored frequently after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproate concentrations drop significantly or seizure control deteriorates [see Drug Interactions (7.1)].
5.17 Effect On Hiv and Cmv Viruses Replication (5.17 Effect on HIV and CMV Viruses Replication)
There are in vitro studies that suggest valproate stimulates the replication of the HIV and CMV viruses under certain experimental conditions. The clinical consequence, if any, is not known. Additionally, the relevance of these in vitro findings is uncertain for patients receiving maximally suppressive antiretroviral therapy. Nevertheless, these data should be borne in mind when interpreting the results from regular monitoring of the viral load in HIV infected patients receiving valproate or when following CMV infected patients clinically.
5.7 Bleeding and Other Hematopoietic Disorders
Valproate is associated with dose-related thrombocytopenia. In a clinical trial of divalproex sodium as monotherapy in patients with epilepsy, 34/126 patients (27%) receiving approximately 50 mg/kg/day on average, had at least one value of platelets ≤ 75 x 109/L. Approximately half of these patients had treatment discontinued, with return of platelet counts to normal. In the remaining patients, platelet counts normalized with continued treatment. In this study, the probability of thrombocytopenia appeared to increase significantly at total valproate concentrations of ≥ 110 mcg/mL (females) or ≥ 135 mcg/mL (males). The therapeutic benefit which may accompany the higher doses should therefore be weighed against the possibility of a greater incidence of adverse effects. Valproate use has also been associated with decreases in other cell lines and myelodysplasia.
Because of reports of cytopenias, inhibition of the secondary phase of platelet aggregation, and abnormal coagulation parameters (e.g., low fibrinogen, coagulation factor deficiencies, acquired von Willebrand's disease), measurements of complete blood counts and coagulation tests are recommended before initiating therapy and at periodic intervals. It is recommended that patients receiving valproate be monitored for blood counts and coagulation parameters prior to planned surgery and during pregnancy [see Use in Specific Populations (8.1)]. Evidence of hemorrhage, bruising, or a disorder of hemostasis/coagulation is an indication for reduction of the dosage or withdrawal of therapy.
5.16 Effect On Ketone and Thyroid Function Tests (5.16 Effect on Ketone and Thyroid Function Tests)
Valproate is partially eliminated in the urine as a keto-metabolite which may lead to a false interpretation of the urine ketone test.
There have been reports of altered thyroid function tests associated with valproate. The clinical significance of these is unknown.
7.1 Effects of Co Administered Drugs On Valproate Clearance (7.1 Effects of Co-Administered Drugs on Valproate Clearance)
Drugs that affect the level of expression of hepatic enzymes, particularly those that elevate levels of glucuronosyltransferases (such as ritonavir), may increase the clearance of valproate. For example, phenytoin, carbamazepine, and phenobarbital (or primidone) can double the clearance of valproate. Thus, patients on monotherapy will generally have longer half-lives and higher concentrations than patients receiving polytherapy with antiepilepsy drugs.
In contrast, drugs that are inhibitors of cytochrome P450 isozymes, e.g., antidepressants, may be expected to have little effect on valproate clearance because cytochrome P450 microsomal mediated oxidation is a relatively minor secondary metabolic pathway compared to glucuronidation and beta-oxidation.
Because of these changes in valproate clearance, monitoring of valproate and concomitant drug concentrations should be increased whenever enzyme inducing drugs are introduced or withdrawn.
The following list provides information about the potential for an influence of several commonly prescribed medications on valproate pharmacokinetics. The list is not exhaustive nor could it be, since new interactions are continuously being reported.
5.9 Hyperammonemia and Encephalopathy Associated With Concomitant Topiramate Use (5.9 Hyperammonemia and Encephalopathy Associated with Concomitant Topiramate Use)
Concomitant administration of topiramate and valproate has been associated with hyperammonemia with or without encephalopathy in patients who have tolerated either drug alone. Clinical symptoms of hyperammonemic encephalopathy often include acute alterations in level of consciousness and/or cognitive function with lethargy or vomiting. Hypothermia can also be a manifestation of hyperammonemia [see Warnings and Precautions (5.10)]. In most cases, symptoms and signs abated with discontinuation of either drug. This adverse reaction is not due to a pharmacokinetic interaction. Patients with inborn errors of metabolism or reduced hepatic mitochondrial activity may be at an increased risk for hyperammonemia with or without encephalopathy. Although not studied, an interaction of topiramate and valproate may exacerbate existing defects or unmask deficiencies in susceptible persons. In patients who develop unexplained lethargy, vomiting, or changes in mental status, hyperammonemic encephalopathy should be considered and an ammonia level should be measured [see Contraindications (4) and Warnings and Precautions (5.8)].
Package Label Principal Display Panel Valproate Sodium 5 Ml Single Dose Vial Label (PACKAGE LABEL - PRINCIPAL DISPLAY PANEL - Valproate Sodium 5 mL Single Dose Vial Label)
NDC 63323-494-41
Valproate
Sodium
Injection, USP
500 mg* per 5 mL
(100 mg per mL)
For intravenous infusion only.
Preservative free.
5 mL Single-dose Vial Rx only
Package Label Principal Display Panel Valproate Sodium 5 Ml Single Dose Vial Tray Label (PACKAGE LABEL - PRINCIPAL DISPLAY PANEL - Valproate Sodium 5 mL Single Dose Vial Tray Label)
NDC 63323-494-16
Valproate
Sodium
Injection, USP
500 mg* per 5 mL
(100 mg per mL)
For intravenous infusion only.
Preservative free.
10 x 5 mL
Single-dose Vials Rx only
5.11 Drug Reaction With Eosinophilia and Systemic Symptoms (dress)/multiorgan Hypersensitivity Reactions (5.11 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity Reactions)
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as Multiorgan Hypersensitivity, has been reported in patients taking valproate. DRESS may be fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy, and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis sometimes resembling an acute viral infection. Eosinophilia is often present. Because this disorder is variable in its expression, other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. Valproate should be discontinued and not be resumed if an alternative etiology for the signs or symptoms cannot be established.
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:45:26.516958 · Updated: 2026-03-14T22:22:25.726819