Zelboraf
38eea320-7e0c-485a-bc30-98c3c45e2763
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
ZELBORAF ® is a kinase inhibitor indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. ( 1.1 , 2.1 ) ZELBORAF ® is indicated for the treatment of patients with Erdheim- Chester Disease with BRAF V600 mutation. ( 1.2 , 2.1 ) Limitation of Use: ZELBORAF is not indicated for treatment of patients with wild-type BRAF melanoma ( 2.1 , 5.2 )
Dosage and Administration
Confirm the presence of BRAF V600E mutation in tumor specimens prior to initiation of treatment with ZELBORAF. ( 2.1 ) Recommended dose: 960 mg orally twice daily taken approximately 12 hours apart with or without a meal. ( 2.2 )
Contraindications
None.
Warnings and Precautions
New Primary Cutaneous Malignancies: Perform dermatologic evaluations prior to initiation of therapy, every 2 months while on therapy, and for up to 6 months following discontinuation of ZELBORAF. Manage with excision and continue treatment without dose adjustment. ( 5.1 ) New Non-Cutaneous Squamous Cell Carcinoma: Evaluate for symptoms or clinical signs of new non-cutaneous SCC before initiation of treatment and periodically during treatment. ( 5.1 ) Other Malignancies: Monitor patients receiving ZELBORAF closely for signs or symptoms of other malignancies ( 5.1 ). Tumor Promotion in BRAF Wild-Type Melanoma: Increased cell proliferation can occur with BRAF inhibitors ( 5.2 ). Serious Hypersensitivity Reactions including anaphylaxis and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS Syndrome): Discontinue ZELBORAF for severe hypersensitivity reactions. ( 5.3 ) Severe Dermatologic Reactions, including Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: Discontinue ZELBORAF for severe dermatologic reactions. ( 5.4 ) QT Prolongation: Monitor ECG and electrolytes before and during treatment. Withhold ZELBORAF for QTc of 500 ms or greater. Correct electrolyte abnormalities and control for cardiac risk factors for QT prolongation. ( 5.5 ) Hepatotoxicity: Measure liver enzymes and bilirubin before initiating ZELBORAF and monitor monthly during treatment. ( 5.6 ) Photosensitivity: Advise patients to avoid sun exposure. ( 5.7 ) Serious Ophthalmologic Reactions: Monitor for signs and symptoms of uveitis. ( 5.8 ) Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of the potential risk to the fetus and to use effective contraception. ( 5.9 , 8.1 , 8.3 ) Radiation Sensitization and Radiation Recall: Severe cases have been reported. ( 5.10 ). Renal Failure: Measure serum creatinine before initiating ZELBORAF and monitor periodically during treatment ( 5.11 ). Dupuytren's Contracture and plantar fascial fibromatosis: Events should be managed with dose reduction, treatment interruption, or treatment discontinuation. ( 5.12 ).
Adverse Reactions
The following adverse reactions are discussed in greater detail in other sections of the label: New Primary Malignancies [see Warnings and Precautions (5.1) ] Hypersensitivity Reactions [see Warnings and Precautions (5.3) ] Dermatologic Reactions [see Warnings and Precautions (5.4) ] QT Prolongation [see Warnings and Precautions (5.5) ] Hepatotoxicity [see Warnings and Precautions (5.6) ] Photosensitivity [see Warnings and Precautions (5.7) ] Ophthalmologic Reactions [see Warnings and Precautions (5.8) ] Radiation Sensitization and Radiation Recall [see Warnings and Precautions (5.10) ] Renal Failure [see Warnings and Precautions (5.11) ] Dupuytren's Contracture and Plantar Fascial Fibromatosis [see Warnings and Precautions (5.12) ]
Drug Interactions
Avoid concomitant administration of ZELBORAF with strong CYP3A4 inhibitors or inducers. ( 7.1 ) CYP1A2 Substrates: ZELBORAF can increase concentrations of CYP1A2 substrates. Avoid concomitant use of ZELBORAF with CYP1A2 substrates with a narrow therapeutic window. If coadministration cannot be avoided, monitor closely for toxicities and consider dose reduction of CYP1A2 substrates. ( 7.2 ).
How Supplied
ZELBORAF (vemurafenib) is supplied as 240 mg film-coated tablets with VEM debossed on one side. The following packaging configurations are available: NDC 50242-090-01 single bottle of 120 count NDC 50242-090-02 single bottle of 112 count
Storage and Handling
ZELBORAF (vemurafenib) is supplied as 240 mg film-coated tablets with VEM debossed on one side. The following packaging configurations are available: NDC 50242-090-01 single bottle of 120 count NDC 50242-090-02 single bottle of 112 count
Description
ZELBORAF ® is a kinase inhibitor indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. ( 1.1 , 2.1 ) ZELBORAF ® is indicated for the treatment of patients with Erdheim- Chester Disease with BRAF V600 mutation. ( 1.2 , 2.1 ) Limitation of Use: ZELBORAF is not indicated for treatment of patients with wild-type BRAF melanoma ( 2.1 , 5.2 )
Medication Information
Warnings and Precautions
New Primary Cutaneous Malignancies: Perform dermatologic evaluations prior to initiation of therapy, every 2 months while on therapy, and for up to 6 months following discontinuation of ZELBORAF. Manage with excision and continue treatment without dose adjustment. ( 5.1 ) New Non-Cutaneous Squamous Cell Carcinoma: Evaluate for symptoms or clinical signs of new non-cutaneous SCC before initiation of treatment and periodically during treatment. ( 5.1 ) Other Malignancies: Monitor patients receiving ZELBORAF closely for signs or symptoms of other malignancies ( 5.1 ). Tumor Promotion in BRAF Wild-Type Melanoma: Increased cell proliferation can occur with BRAF inhibitors ( 5.2 ). Serious Hypersensitivity Reactions including anaphylaxis and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS Syndrome): Discontinue ZELBORAF for severe hypersensitivity reactions. ( 5.3 ) Severe Dermatologic Reactions, including Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: Discontinue ZELBORAF for severe dermatologic reactions. ( 5.4 ) QT Prolongation: Monitor ECG and electrolytes before and during treatment. Withhold ZELBORAF for QTc of 500 ms or greater. Correct electrolyte abnormalities and control for cardiac risk factors for QT prolongation. ( 5.5 ) Hepatotoxicity: Measure liver enzymes and bilirubin before initiating ZELBORAF and monitor monthly during treatment. ( 5.6 ) Photosensitivity: Advise patients to avoid sun exposure. ( 5.7 ) Serious Ophthalmologic Reactions: Monitor for signs and symptoms of uveitis. ( 5.8 ) Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of the potential risk to the fetus and to use effective contraception. ( 5.9 , 8.1 , 8.3 ) Radiation Sensitization and Radiation Recall: Severe cases have been reported. ( 5.10 ). Renal Failure: Measure serum creatinine before initiating ZELBORAF and monitor periodically during treatment ( 5.11 ). Dupuytren's Contracture and plantar fascial fibromatosis: Events should be managed with dose reduction, treatment interruption, or treatment discontinuation. ( 5.12 ).
Indications and Usage
ZELBORAF ® is a kinase inhibitor indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. ( 1.1 , 2.1 ) ZELBORAF ® is indicated for the treatment of patients with Erdheim- Chester Disease with BRAF V600 mutation. ( 1.2 , 2.1 ) Limitation of Use: ZELBORAF is not indicated for treatment of patients with wild-type BRAF melanoma ( 2.1 , 5.2 )
Dosage and Administration
Confirm the presence of BRAF V600E mutation in tumor specimens prior to initiation of treatment with ZELBORAF. ( 2.1 ) Recommended dose: 960 mg orally twice daily taken approximately 12 hours apart with or without a meal. ( 2.2 )
Contraindications
None.
Adverse Reactions
The following adverse reactions are discussed in greater detail in other sections of the label: New Primary Malignancies [see Warnings and Precautions (5.1) ] Hypersensitivity Reactions [see Warnings and Precautions (5.3) ] Dermatologic Reactions [see Warnings and Precautions (5.4) ] QT Prolongation [see Warnings and Precautions (5.5) ] Hepatotoxicity [see Warnings and Precautions (5.6) ] Photosensitivity [see Warnings and Precautions (5.7) ] Ophthalmologic Reactions [see Warnings and Precautions (5.8) ] Radiation Sensitization and Radiation Recall [see Warnings and Precautions (5.10) ] Renal Failure [see Warnings and Precautions (5.11) ] Dupuytren's Contracture and Plantar Fascial Fibromatosis [see Warnings and Precautions (5.12) ]
Drug Interactions
Avoid concomitant administration of ZELBORAF with strong CYP3A4 inhibitors or inducers. ( 7.1 ) CYP1A2 Substrates: ZELBORAF can increase concentrations of CYP1A2 substrates. Avoid concomitant use of ZELBORAF with CYP1A2 substrates with a narrow therapeutic window. If coadministration cannot be avoided, monitor closely for toxicities and consider dose reduction of CYP1A2 substrates. ( 7.2 ).
Storage and Handling
ZELBORAF (vemurafenib) is supplied as 240 mg film-coated tablets with VEM debossed on one side. The following packaging configurations are available: NDC 50242-090-01 single bottle of 120 count NDC 50242-090-02 single bottle of 112 count
How Supplied
ZELBORAF (vemurafenib) is supplied as 240 mg film-coated tablets with VEM debossed on one side. The following packaging configurations are available: NDC 50242-090-01 single bottle of 120 count NDC 50242-090-02 single bottle of 112 count
Description
ZELBORAF ® is a kinase inhibitor indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. ( 1.1 , 2.1 ) ZELBORAF ® is indicated for the treatment of patients with Erdheim- Chester Disease with BRAF V600 mutation. ( 1.2 , 2.1 ) Limitation of Use: ZELBORAF is not indicated for treatment of patients with wild-type BRAF melanoma ( 2.1 , 5.2 )
Section 42229-5
Limitation of Use: ZELBORAF is not indicated for treatment of patients with wild-type BRAF melanoma [see Warnings and Precautions (5.2)].
Section 42231-1
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: 11/2017 | |
|
MEDICATION GUIDE |
||
|
What is the most important information I should know about ZELBORAF? ZELBORAF can cause serious side effects, including: Risk of new cancers. ZELBORAF may cause certain types of skin cancer called cutaneous squamous cell carcinoma (cuSCC) and keratoacanthoma. New melanoma lesions have occurred in people who take ZELBORAF. ZELBORAF may also cause another type of cancer called non-cutaneous squamous cell carcinoma (non-cuSCC). Talk with your healthcare provider about your risk for these cancers. Check your skin and tell your healthcare provider right away about any skin changes including a:
Your healthcare provider should check your skin before you start taking ZELBORAF, and every 2 months during treatment with ZELBORAF, to look for any new skin cancers. Your healthcare provider may continue to check your skin for 6 months after you stop taking ZELBORAF. Your healthcare provider should also check for cancers that may not occur on the skin. Tell your healthcare provider about any new symptoms that you get while taking ZELBORAF. Other blood cell cancers have happened in some people with Erdheim-Chester Disease (ECD) including those who take ZELBORAF. If you have other blood cell cancers and take ZELBORAF for ECD, your healthcare provider will monitor your blood cancer through routine blood tests. See "What are the possible side effects of ZELBORAF?" for more information about side effects. |
||
|
What is ZELBORAF? ZELBORAF is a prescription medicine used to treat:
ZELBORAF is not used to treat melanoma with a normal BRAF gene. Your healthcare provider will perform a test to make sure that ZELBORAF is right for you.
It is not known if ZELBORAF is safe and effective in children under 18 years of age. |
||
|
Before you take ZELBORAF, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
||
|
How should I take ZELBORAF?
|
||
|
What should I avoid while taking ZELBORAF? Avoid sunlight during treatment with ZELBORAF. ZELBORAF can make your skin sensitive to sunlight. You may burn more easily and get severe sunburns. To help protect against sunburn:
|
||
|
What are the possible side effects of ZELBORAF? ZELBORAF may cause serious side effects, including:
|
||
|
|
|
|
||
|
|
|
The most common side effects of ZELBORAF in melanoma include: |
||
|
|
|
|
The most common side effects of ZELBORAF in Erdheim-Chester Disease include: |
||
|
|
|
These are not all the possible side effects of ZELBORAF. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Genentech at 1-888-835-2555. |
||
|
How should I store ZELBORAF?
Keep ZELBORAF and all medicine out of the reach of children. |
||
|
General information about the safe and effective use of ZELBORAF. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ZELBORAF for a condition for which it was not prescribed. Do not give ZELBORAF to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ZELBORAF that is written for health professionals. |
||
|
What are the ingredients in ZELBORAF? Active ingredient: vemurafenib Inactive ingredients: Tablet Core: hypromellose acetate succinate, croscarmellose sodium, colloidal silicon dioxide, magnesium stearate, and hydroxypropyl cellulose. Coating: pinkish white: poly (vinyl alcohol), titanium dioxide, polyethylene glycol 3350, talc, and iron oxide red. Distributed by: Genentech USA, Inc., A Member of the Roche Group,1 DNA Way, South San Francisco, CA 94080-4990 |
Section 44425-7
Storage and Stability: Store at room temperature 20°C–25°C (68°F–77°F); excursions permitted between 15°C and 30°C (59°F and 86°F), See USP Controlled Room Temperature. Store in the original container with the lid tightly closed.
10 Overdosage
There is no information on overdosage of ZELBORAF.
8.2 Lactation
There is no information available regarding the presence of vemurafenib in human milk, effects on the breastfed infant, or effects on milk production. Because of the potential for serious adverse reactions in a breastfed infant, including malignancy, severe dermatologic reactions, QT prolongation, hepatotoxicity, photosensitivity, and ophthalmologic toxicity, [see Warnings and Precautions (5)], advise women not to breastfeed during treatment with ZELBORAF and for 2 weeks after the final dose.
11 Description
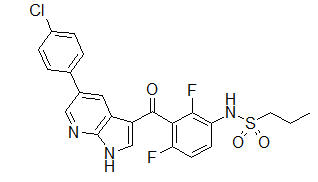
ZELBORAF (vemurafenib) is a kinase inhibitor available as 240 mg tablets for oral use. Vemurafenib has the chemical name propane-1-sulfonic acid {3-[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]- 2,4-difluoro-phenyl}-amide. It has the molecular formula C23H18ClF2N3O3S and a molecular weight of 489.9. Vemurafenib has the following chemical structure:
Vemurafenib is a white to off-white crystalline solid. It is practically insoluble in aqueous media.
Tablets of ZELBORAF are for oral administration. Each tablet contains 240 mg of vemurafenib.
The inactive ingredients of ZELBORAF are: Tablet core: hypromellose acetate succinate, croscarmellose sodium, colloidal silicon dioxide, magnesium stearate, and hydroxypropyl cellulose. Coating: pinkish white: poly (vinyl alcohol), titanium dioxide, polyethylene glycol 3350, talc, and iron oxide red.
8.4 Pediatric Use
The safety and effectiveness of ZELBORAF in pediatric patients have not been established. Vemurafenib was studied in 6 adolescent patients 15 to 17 years of age with unresectable or metastatic melanoma with BRAF V600 mutation. A maximum tolerated dose was not reached with doses up to vemurafenib 960 mg twice daily. No new safety signals were observed. Vemurafenib steady-state exposure in these 6 adolescent patients was generally similar to that in adults.
8.5 Geriatric Use
Clinical studies of ZELBORAF did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
5.11 Renal Failure
Renal failure, including acute interstitial nephritis and acute tubular necrosis, can occur with ZELBORAF. In Trial 1, in patients with metastatic melanoma, 26% of ZELBORAF-treated patients and 5% of dacarbazine-treated patients experienced Grade 1-2 creatinine elevations [greater than 1 and up to 3 times upper limit of normal (ULN)]; 1.2% of ZELBORAF-treated patients and 1.1% of dacarbazine-treated patients experienced Grade 3-4 creatinine elevations (greater than 3 times ULN).
In Trial 4, in patients with ECD, 86% (19/22) of patients experienced Grade 1/2 creatinine elevations and 9.1% (2/22) of patients experienced Grade 3 creatinine elevations.
Measure serum creatinine before initiation of ZELBORAF and periodically during treatment.
5.6 Hepatotoxicity
Liver injury leading to functional hepatic impairment, including coagulopathy or other organ dysfunction, can occur with ZELBORAF [see Adverse Reactions (6.1)]. Monitor transaminases, alkaline phosphatase, and bilirubin before initiation of treatment and monthly during treatment, or as clinically indicated. Manage laboratory abnormalities with dose reduction, treatment interruption, or treatment discontinuation [see Dosage and Administration (2.3)].
4 Contraindications
None.
5.5 Qt Prolongation
Concentration-dependent QT prolongation occurred in an uncontrolled, open-label QT sub-study in previously treated patients with BRAF V600E mutation-positive metastatic melanoma [see Clinical Pharmacology (12.2)]. QT prolongation may lead to an increased risk of ventricular arrhythmias, including Torsade de Pointes.
Do not start treatment in patients with uncorrectable electrolyte abnormalities, QTc > 500 ms, or long QT syndrome, or in patients who are taking medicinal products known to prolong the QT interval. Prior to and following treatment initiation or after dose modification of ZELBORAF for QTc prolongation, evaluate ECG and electrolytes (including potassium, magnesium, and calcium) after 15 days, monthly during the first 3 months, and then every 3 months thereafter or more often as clinically indicated.
Withhold ZELBORAF in patients who develop QTc > 500 ms (Grade 3). Upon recovery to QTc ≤ 500 ms (Grade ≤ 2), restart at a reduced dose. Permanently discontinue ZELBORAF treatment if the QTc interval remains > 500 ms and increased > 60 ms from pre-treatment values after controlling cardiac risk factors for QT prolongation (e.g., electrolyte abnormalities, congestive heart failure, and bradyarrhythmias) [see Dosage and Administration (2.3)].
6 Adverse Reactions
The following adverse reactions are discussed in greater detail in other sections of the label:
- New Primary Malignancies [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Dermatologic Reactions [see Warnings and Precautions (5.4)]
- QT Prolongation [see Warnings and Precautions (5.5)]
- Hepatotoxicity [see Warnings and Precautions (5.6)]
- Photosensitivity [see Warnings and Precautions (5.7)]
- Ophthalmologic Reactions [see Warnings and Precautions (5.8)]
- Radiation Sensitization and Radiation Recall [see Warnings and Precautions (5.10)]
- Renal Failure [see Warnings and Precautions (5.11)]
- Dupuytren's Contracture and Plantar Fascial Fibromatosis [see Warnings and Precautions (5.12)]
7 Drug Interactions
- Avoid concomitant administration of ZELBORAF with strong CYP3A4 inhibitors or inducers. (7.1)
- CYP1A2 Substrates: ZELBORAF can increase concentrations of CYP1A2 substrates. Avoid concomitant use of ZELBORAF with CYP1A2 substrates with a narrow therapeutic window. If coadministration cannot be avoided, monitor closely for toxicities and consider dose reduction of CYP1A2 substrates. (7.2).
2.2 Recommended Dose
The recommended dose of ZELBORAF is 960 mg (four 240 mg tablets) orally every 12 hours with or without a meal. A missed dose can be taken up to 4 hours prior to the next dose.
Treat patients with ZELBORAF until disease progression or unacceptable toxicity occurs.
Do not take an additional dose if vomiting occurs after ZELBORAF administration, but continue with the next scheduled dose.
Do not crush or chew the tablets.
5.7 Photosensitivity
Mild to severe photosensitivity can occur in patients treated with ZELBORAF [see Adverse Reactions (6.1)]. Advise patients to avoid sun exposure, wear protective clothing and use a broad spectrum UVA/UVB sunscreen and lip balm (SPF ≥ 30) when outdoors.
Institute dose modifications for intolerable Grade 2 or greater photosensitivity [see Dosage and Administration (2.2)].
8.7 Renal Impairment
No formal clinical study has been conducted to evaluate the effect of renal impairment on the pharmacokinetics of vemurafenib. No dose adjustment is recommended for patients with mild and moderate renal impairment based on a population pharmacokinetic analysis [see Clinical Pharmacology (12.3)]. The appropriate dose of ZELBORAF has not been established in patients with severe renal impairment.
12.3 Pharmacokinetics
The pharmacokinetics of vemurafenib were determined in patients with BRAF mutation-positive metastatic melanoma following 15 days of 960 mg twice daily with dosing approximately 12 hours apart. The population pharmacokinetic analysis pooled data from 458 patients. At steady-state, vemurafenib exhibits linear pharmacokinetics within the 240 mg to 960 mg dose range.
8.6 Hepatic Impairment
No formal clinical study has been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of vemurafenib. No dose adjustment is recommended for patients with mild and moderate hepatic impairment based on a population pharmacokinetic analysis [see Clinical Pharmacology (12.3)]. The appropriate dose of ZELBORAF has not been established in patients with severe hepatic impairment.
1 Indications and Usage
- ZELBORAF ® is a kinase inhibitor indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. (1.1, 2.1)
- ZELBORAF® is indicated for the treatment of patients with Erdheim- Chester Disease with BRAF V600 mutation. (1.2, 2.1)
Limitation of Use: ZELBORAF is not indicated for treatment of patients with wild-type BRAF melanoma (2.1, 5.2)
12.1 Mechanism of Action
Vemurafenib is a low molecular weight, orally available inhibitor of some mutated forms of BRAF serine- threonine kinase, including BRAF V600E. Vemurafenib also inhibits other kinases in vitro such as CRAF, ARAF, wild-type BRAF, SRMS, ACK1, MAP4K5, and FGR at similar concentrations. Some mutations in the BRAF gene including V600E result in constitutively activated BRAF proteins, which can cause cell proliferation in the absence of growth factors that would normally be required for proliferation. Vemurafenib has anti-tumor effects in cellular and animal models of melanomas with mutated BRAF V600E.
5.9 Embryo Fetal Toxicity
Based on its mechanism of action, ZELBORAF can cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ZELBORAF and for 2 weeks after the final dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
7.3 Concurrent Ipilimumab
Increases in transaminases and bilirubin occurred in a majority of patients who received concurrent ipilimumab and ZELBORAF [see Warnings and Precautions Section 5.6].
5 Warnings and Precautions
- New Primary Cutaneous Malignancies: Perform dermatologic evaluations prior to initiation of therapy, every 2 months while on therapy, and for up to 6 months following discontinuation of ZELBORAF. Manage with excision and continue treatment without dose adjustment. (5.1)
- New Non-Cutaneous Squamous Cell Carcinoma: Evaluate for symptoms or clinical signs of new non-cutaneous SCC before initiation of treatment and periodically during treatment. (5.1)
- Other Malignancies: Monitor patients receiving ZELBORAF closely for signs or symptoms of other malignancies (5.1).
- Tumor Promotion in BRAF Wild-Type Melanoma: Increased cell proliferation can occur with BRAF inhibitors (5.2).
- Serious Hypersensitivity Reactions including anaphylaxis and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS Syndrome): Discontinue ZELBORAF for severe hypersensitivity reactions. (5.3)
- Severe Dermatologic Reactions, including Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: Discontinue ZELBORAF for severe dermatologic reactions. (5.4)
- QT Prolongation: Monitor ECG and electrolytes before and during treatment. Withhold ZELBORAF for QTc of 500 ms or greater. Correct electrolyte abnormalities and control for cardiac risk factors for QT prolongation. (5.5)
- Hepatotoxicity: Measure liver enzymes and bilirubin before initiating ZELBORAF and monitor monthly during treatment. (5.6)
- Photosensitivity: Advise patients to avoid sun exposure. (5.7)
- Serious Ophthalmologic Reactions: Monitor for signs and symptoms of uveitis. (5.8)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of the potential risk to the fetus and to use effective contraception. (5.9, 8.1, 8.3)
- Radiation Sensitization and Radiation Recall: Severe cases have been reported. (5.10).
- Renal Failure: Measure serum creatinine before initiating ZELBORAF and monitor periodically during treatment (5.11).
- Dupuytren's Contracture and plantar fascial fibromatosis: Events should be managed with dose reduction, treatment interruption, or treatment discontinuation. (5.12).
5.4 Dermatologic Reactions
Severe dermatologic reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis, can occur in patients receiving ZELBORAF. Permanently discontinue ZELBORAF in patients who experience a severe dermatologic reaction [see Adverse Reactions (6.1)].
1.2 Erdheim Chester Disease
ZELBORAF® is indicated for the treatment of patients with Erdheim-Chester Disease (ECD) with BRAF V600 mutation.
2 Dosage and Administration
3 Dosage Forms and Strengths
Tablet: 240 mg.
5.8 Ophthalmologic Reactions
Uveitis, blurry vision, and photophobia can occur in patients treated with ZELBORAF. In Trial 1, uveitis, including iritis, occurred in 2.1% (7/336) of patients receiving ZELBORAF compared to no patients in the dacarbazine arm. Treatment with steroid and mydriatic ophthalmic drops may be required to manage uveitis. Monitor patients for signs and symptoms of uveitis.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ZELBORAF. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Neoplasms benign, malignant and unspecified (incl. cysts and polyps): Progression of pre-existing chronic myelomonocytic leukemia with NRAS mutation [see Warnings and Precautions (5.1)].
Skin and subcutaneous tissue disorders: Drug reaction with eosinophilia and systemic symptoms (DRESS syndrome) [see Warnings and Precautions (5.3)].
Blood and lymphatic systems disorder: Neutropenia
Injury, poisoning and procedural complications: Radiation sensitization and recall [see Warnings and Precautions (5.10)].
Gastrointestinal disorders: Pancreatitis
Renal and urinary disorders: Acute interstitial nephritis, acute tubular necrosis [see Warnings and Precautions (5.11)].
Musculoskeletal and connective tissue disorders: Dupuytren's contracture and plantar fascial fibromatosis [see Warnings and Precautions (5.12)].
8 Use in Specific Populations
- Lactation: Do not breastfeed while taking ZELBORAF. (8.2)
5.3 Hypersensitivity Reactions
Anaphylaxis and other serious hypersensitivity reactions can occur during treatment and upon re-initiation of treatment with ZELBORAF. Severe hypersensitivity reactions included generalized rash and erythema, hypotension, and drug reaction with eosinophilia and systemic symptoms (DRESS syndrome). Permanently discontinue ZELBORAF in patients who experience a severe hypersensitivity reaction [see Adverse Reactions (6.2)].
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
Unresectable or Metastatic Melanoma with BRAF V600E Mutation This section describes adverse drug reactions (ADRs) identified from analyses of Trial 1 and Trial 2 [see Clinical Studies (14)]. Trial 1 randomized (1:1) 675 treatment-naive patients with unresectable or metastatic melanoma to receive ZELBORAF 960 mg orally twice daily or dacarbazine 1000 mg/m2 intravenously every 3 weeks. In Trial 2, 132 patients with metastatic melanoma and failure of at least one prior systemic therapy received treatment with ZELBORAF 960 mg orally twice daily.
Table 1 presents adverse reactions reported in at least 10% of unresectable or metastatic melanoma patients treated with ZELBORAF. The most common adverse reactions of any grade (≥ 30% in either study) in ZELBORAF-treated patients were arthralgia, rash, alopecia, fatigue, photosensitivity reaction, nausea, pruritus, and skin papilloma. The most common (≥ 5%) Grade 3 adverse reactions were cuSCC and rash. The incidence of Grade 4 adverse reactions was ≤ 4% in both studies.
The incidence of adverse events resulting in permanent discontinuation of study medication in Trial 1 was 7% for the ZELBORAF arm and 4% for the dacarbazine arm. In Trial 2, the incidence of adverse events resulting in permanent discontinuation of study medication was 3% in ZELBORAF-treated patients. The median duration of study treatment was 4.2 months for ZELBORAF and 0.8 months for dacarbazine in Trial 1, and 5.7 months for ZELBORAF in Trial 2.
| ADRs | Trial 1: Treatment-Naïve Patients | Trial 2: Patients with Failure of at Least One Prior Systemic Therapy | ||||
|---|---|---|---|---|---|---|
| ZELBORAF n=336 |
Dacarbazine n=287 |
ZELBORAF n=132 |
||||
| All Grades (%) |
Grade 3 Grade 4 adverse reactions limited to gamma-glutamyltransferase increased (< 1% in Trial 1 and 4% in Trial 2).
(%) |
All Grades (%) |
Grade 3 (%) |
All Grades (%) | Grade 3 (%) | |
| Skin and subcutaneous tissue disorders | ||||||
| Rash | 37 | 8 | 2 | 0 | 52 | 7 |
| Photosensitivity reaction | 33 | 3 | 4 | 0 | 49 | 3 |
| Alopecia | 45 | < 1 | 2 | 0 | 36 | 0 |
| Pruritus | 23 | 1 | 1 | 0 | 30 | 2 |
| Hyperkeratosis | 24 | 1 | < 1 | 0 | 28 | 0 |
| Rash maculo-papular | 9 | 2 | < 1 | 0 | 21 | 6 |
| Actinic keratosis | 8 | 0 | 3 | 0 | 17 | 0 |
| Dry skin | 19 | 0 | 1 | 0 | 16 | 0 |
| Rash papular | 5 | < 1 | 0 | 0 | 13 | 0 |
| Erythema | 14 | 0 | 2 | 0 | 8 | 0 |
| Musculoskeletal and connective tissue disorders | ||||||
| Arthralgia | 53 | 4 | 3 | < 1 | 67 | 8 |
| Myalgia | 13 | < 1 | 1 | 0 | 24 | < 1 |
| Pain in extremity | 18 | < 1 | 6 | 2 | 9 | 0 |
| Musculoskeletal pain | 8 | 0 | 4 | < 1 | 11 | 0 |
| Back pain | 8 | < 1 | 5 | < 1 | 11 | < 1 |
| General disorders and administration site conditions | ||||||
| Fatigue | 38 | 2 | 33 | 2 | 54 | 4 |
| Edema peripheral | 17 | < 1 | 5 | 0 | 23 | 0 |
| Pyrexia | 19 | < 1 | 9 | < 1 | 17 | 2 |
| Asthenia | 11 | < 1 | 9 | < 1 | 2 | 0 |
| Gastrointestinal disorders | ||||||
| Nausea | 35 | 2 | 43 | 2 | 37 | 2 |
| Diarrhea | 28 | < 1 | 13 | < 1 | 29 | < 1 |
| Vomiting | 18 | 1 | 26 | 1 | 26 | 2 |
| Constipation | 12 | < 1 | 24 | 0 | 16 | 0 |
| Nervous system disorders | ||||||
| Headache | 23 | < 1 | 10 | 0 | 27 | 0 |
| Dysgeusia | 14 | 0 | 3 | 0 | 11 | 0 |
| Neoplasms benign, malignant and unspecified (includes cysts and polyps) | ||||||
| Skin papilloma | 21 | < 1 | 0 | 0 | 30 | 0 |
| Cutaneous SCC Includes both squamous cell carcinoma of the skin and keratoacanthoma.
Cases of cutaneous squamous cell carcinoma were required to be reported as Grade 3 per protocol.
|
24 | 22 | < 1 | < 1 | 24 | 24 |
| Seborrheic keratosis | 10 | < 1 | 1 | 0 | 14 | 0 |
| Investigations | ||||||
| Gamma-glutamyltransferase increased | 5 | 3 | 1 | 0 | 15 | 6 |
| Metabolism and nutrition disorders | ||||||
| Decreased appetite | 18 | 0 | 8 | < 1 | 21 | 0 |
| Respiratory, thoracic and mediastinal disorders | ||||||
| Cough | 8 | 0 | 7 | 0 | 12 | 0 |
| Injury, poisoning and procedural complications | ||||||
| Sunburn | 10 | 0 | 0 | 0 | 14 | 0 |
Clinically relevant adverse reactions reported in < 10% of unresectable or metastatic melanoma patients treated with ZELBORAF in the Phase 2 and Phase 3 studies include:
Skin and subcutaneous tissue disorders: palmar-plantar erythrodysesthesia syndrome, keratosis pilaris, panniculitis, erythema nodosum, Stevens-Johnson syndrome, toxic epidermal necrolysis
Musculoskeletal and connective tissue disorders: arthritis, Dupuytren's contracture
Nervous system disorders: neuropathy peripheral, VIIth nerve paralysis
Neoplasms benign, malignant and unspecified (includes cysts and polyps): basal cell carcinoma, oropharyngeal squamous cell carcinoma
Infections and infestations: folliculitis
Eye disorders: retinal vein occlusion
Vascular disorders: vasculitis
Cardiac disorders: atrial fibrillation
Table 2 shows the incidence of worsening liver laboratory abnormalities in Trial 1 summarized as the proportion of patients who experienced a shift from baseline to Grade 3 or 4.
| Parameter | Change From Baseline to Grade 3/4 | |
|---|---|---|
| ZELBORAF (%) | Dacarbazine (%) | |
| GGT | 11.5 | 8.6 |
| AST | 0.9 | 0.4 |
| ALT | 2.8 | 1.9 |
| Alkaline phosphatase | 2.9 | 0.4 |
| Bilirubin | 1.9 | 0 |
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Healthcare providers should advise patients of the potential benefits and risks of ZELBORAF and instruct their patients to read the Medication Guide before starting ZELBORAF therapy. Inform patients of the following:
- Evidence of BRAF V600E mutation in the tumor specimen with an FDA approved test is necessary to identify patients with melanoma for whom treatment with ZELBORAF is indicated [see Dosage and Administration (2.1)].
- ZELBORAF increases the risk of developing new primary cutaneous malignancies. Advise patients of the importance of contacting their healthcare provider immediately for any changes in their skin [see Warnings and Precautions (5.1)].
- Anaphylaxis and other serious hypersensitivity reactions can occur during treatment and upon re- initiation of treatment with ZELBORAF. Advise patients to stop taking ZELBORAF and to seek immediate medical attention for symptoms of anaphylaxis or hypersensitivity [see Warnings and Precautions (5.3)].
- Severe dermatologic reactions can occur in patients receiving ZELBORAF. Advise patients to stop taking ZELBORAF and to contact their health-care provider for severe dermatologic reactions [see Warnings and Precautions (5.4)].
- ZELBORAF can prolong QT interval, which may result in ventricular arrhythmias. Advise patients of the importance of monitoring of their electrolytes and the electrical activity of their heart (via an ECG) during ZELBORAF treatment [see Warnings and Precautions (5.5)].
- Liver injury leading to functional hepatic impairment, including coagulopathy or other organ dysfunction, can occur with ZELBORAF. Advise patients of the importance of laboratory monitoring of their liver during ZELBORAF treatment and to contact their health-care provider for relevant symptoms [see Warnings and Precautions (5.6)].
- ZELBORAF can cause mild to severe photosensitivity. Advise patients to avoid sun exposure, wear protective clothing, and use a broad spectrum UVA/UVB sunscreen and lip balm (SPF ≥ 30) when outdoors to help protect against sunburn [see Warnings and Precautions (5.7)].
- Ophthalmologic reactions can occur in patients treated with ZELBORAF. Advise patients to contact their health-care provider immediately for ophthalmologic symptoms [see Warnings and Precautions (5.8)].
16 How Supplied/storage and Handling
ZELBORAF (vemurafenib) is supplied as 240 mg film-coated tablets with VEM debossed on one side. The following packaging configurations are available:
NDC 50242-090-01 single bottle of 120 count
NDC 50242-090-02 single bottle of 112 count
1.1 Unresectable Or Metastatic Melanoma
ZELBORAF ® is indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
13.2 Animal Toxicology And/or Pharmacology
Consistent with the increased incidence of cutaneous squamous cell carcinomas in patients treated with vemurafenib, the treatment of mice implanted with human cuSCC cells with vemurafenib caused a dose- dependent acceleration of the growth of the implanted tumors.
7.4 Effect of Vemurafenib On P Gp Substrates
Coadministration of ZELBORAF with digoxin, a sensitive P-glycoprotein (P-gp) substrate, increased digoxin systemic exposure by 1.8-fold. Avoid concurrent use of P-gp substrates known to have narrow therapeutic indices. If use of these medications is unavoidable, consider dose reduction of P-gp substrates with narrow therapeutic indices.
5.2 Tumor Promotion in Braf Wild Type Melanoma
In vitro experiments have demonstrated paradoxical activation of MAP-kinase signaling and increased cell proliferation in BRAF wild-type cells that are exposed to BRAF inhibitors. Confirm evidence of BRAF V600E mutation in tumor specimens prior to initiation of ZELBORAF [see Indications and Usage (1) and Dosage and Administration (2.1)].
7.2 Effect of Vemurafenib On Cyp1a2 Substrates
Coadministration of ZELBORAF with tizanidine, a sensitive CYP1A2 substrate, increased tizanidine systemic exposure by 4.7-fold. Avoid concomitant use of ZELBORAF with drugs having a narrow therapeutic window that are predominantly metabolized by CYP1A2 [see Clinical Pharmacology (12.3)]. If coadministration cannot be avoided, monitor closely for toxicities and consider a dose reduction of concomitant CYP1A2 substrates.
2.1 Patient Selection for Treatment of Melanoma
Confirm the presence of BRAF V600E mutation in melanoma tumor specimens prior to initiation of treatment with ZELBORAF [see Warnings and Precautions (5.2)]. Information on FDA-approved tests for the detection of BRAF V600 mutations in melanoma is available at http://www.fda.gov/CompanionDiagnostics.
2.4 Dose Modification for Strong Cyp3a4 Inducers
Avoid concomitant use of strong CYP3A4 inducers during treatment with ZELBORAF [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]. If concomitant use of a strong CYP3A4 inducer is unavoidable, increase the dose of ZELBORAF by 240 mg (one tablet) as tolerated. After discontinuation of a strong CYP3A4 inducer for two weeks, resume the ZELBORAF dose that was taken prior to initiating the strong CYP3A4 inducer.
5.10 Radiation Sensitization and Radiation Recall
Radiation sensitization and recall, in some cases severe, involving cutaneous and visceral organs have been reported in patients treated with radiation prior to, during, or subsequent to vemurafenib treatment. Fatal cases have been reported in patients with visceral organ involvement. [see Adverse Reactions (6.2)].
Monitor patients closely when vemurafenib is administered concomitantly or sequentially with radiation treatment.
Principal Display Panel 240 Mg Tablet Bottle Carton
NDC 50242-090-02
Zelboraf®
(vemurafenib)
tablets
240 mg
Do not crush or chew tablet.
Rx only
Attention Pharmacist: Dispense the
accompanying Medication Guide to
each patient.
112 tablets
Genentech | Daiichi Sankyo, Inc.
10210025
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
There have been no formal studies conducted assessing the carcinogenic potential of vemurafenib. ZELBORAF increased the development of cutaneous squamous cell carcinomas in patients in clinical trials.
Vemurafenib did not cause genetic damage when tested in in vitro assays (bacterial mutation [AMES Assay], human lymphocyte chromosome aberration) or in the in vivo rat bone marrow micronucleus test.
No specific studies with vemurafenib have been conducted in animals to evaluate the effect on fertility; nevertheless, no histopathological findings were noted in reproductive organs in males and females in repeat-dose toxicology studies in rats at doses up to 450 mg/kg/day (approximately 0.6 and 1.6 times the human exposure based on AUC in males and females, respectively) and dogs at doses up to 450 mg/kg/day (approximately 0.3 times the human clinical exposure based on AUC in both males and females, respectively).
5.12 Dupuytren's Contracture and Plantar Fascial Fibromatosis
Dupuytren's contracture and plantar fascial fibromatosis have been reported with ZELBORAF. The majority of cases were mild to moderate, but severe, disabling cases of Dupuytren's contracture have also been reported [see Dosage and Administration (2.3), Adverse Reactions (6.1, 6.2)].
Structured Label Content
Section 42229-5 (42229-5)
Limitation of Use: ZELBORAF is not indicated for treatment of patients with wild-type BRAF melanoma [see Warnings and Precautions (5.2)].
Section 42231-1 (42231-1)
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: 11/2017 | |
|
MEDICATION GUIDE |
||
|
What is the most important information I should know about ZELBORAF? ZELBORAF can cause serious side effects, including: Risk of new cancers. ZELBORAF may cause certain types of skin cancer called cutaneous squamous cell carcinoma (cuSCC) and keratoacanthoma. New melanoma lesions have occurred in people who take ZELBORAF. ZELBORAF may also cause another type of cancer called non-cutaneous squamous cell carcinoma (non-cuSCC). Talk with your healthcare provider about your risk for these cancers. Check your skin and tell your healthcare provider right away about any skin changes including a:
Your healthcare provider should check your skin before you start taking ZELBORAF, and every 2 months during treatment with ZELBORAF, to look for any new skin cancers. Your healthcare provider may continue to check your skin for 6 months after you stop taking ZELBORAF. Your healthcare provider should also check for cancers that may not occur on the skin. Tell your healthcare provider about any new symptoms that you get while taking ZELBORAF. Other blood cell cancers have happened in some people with Erdheim-Chester Disease (ECD) including those who take ZELBORAF. If you have other blood cell cancers and take ZELBORAF for ECD, your healthcare provider will monitor your blood cancer through routine blood tests. See "What are the possible side effects of ZELBORAF?" for more information about side effects. |
||
|
What is ZELBORAF? ZELBORAF is a prescription medicine used to treat:
ZELBORAF is not used to treat melanoma with a normal BRAF gene. Your healthcare provider will perform a test to make sure that ZELBORAF is right for you.
It is not known if ZELBORAF is safe and effective in children under 18 years of age. |
||
|
Before you take ZELBORAF, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
||
|
How should I take ZELBORAF?
|
||
|
What should I avoid while taking ZELBORAF? Avoid sunlight during treatment with ZELBORAF. ZELBORAF can make your skin sensitive to sunlight. You may burn more easily and get severe sunburns. To help protect against sunburn:
|
||
|
What are the possible side effects of ZELBORAF? ZELBORAF may cause serious side effects, including:
|
||
|
|
|
|
||
|
|
|
The most common side effects of ZELBORAF in melanoma include: |
||
|
|
|
|
The most common side effects of ZELBORAF in Erdheim-Chester Disease include: |
||
|
|
|
These are not all the possible side effects of ZELBORAF. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Genentech at 1-888-835-2555. |
||
|
How should I store ZELBORAF?
Keep ZELBORAF and all medicine out of the reach of children. |
||
|
General information about the safe and effective use of ZELBORAF. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ZELBORAF for a condition for which it was not prescribed. Do not give ZELBORAF to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ZELBORAF that is written for health professionals. |
||
|
What are the ingredients in ZELBORAF? Active ingredient: vemurafenib Inactive ingredients: Tablet Core: hypromellose acetate succinate, croscarmellose sodium, colloidal silicon dioxide, magnesium stearate, and hydroxypropyl cellulose. Coating: pinkish white: poly (vinyl alcohol), titanium dioxide, polyethylene glycol 3350, talc, and iron oxide red. Distributed by: Genentech USA, Inc., A Member of the Roche Group,1 DNA Way, South San Francisco, CA 94080-4990 |
Section 44425-7 (44425-7)
Storage and Stability: Store at room temperature 20°C–25°C (68°F–77°F); excursions permitted between 15°C and 30°C (59°F and 86°F), See USP Controlled Room Temperature. Store in the original container with the lid tightly closed.
10 Overdosage (10 OVERDOSAGE)
There is no information on overdosage of ZELBORAF.
8.2 Lactation
There is no information available regarding the presence of vemurafenib in human milk, effects on the breastfed infant, or effects on milk production. Because of the potential for serious adverse reactions in a breastfed infant, including malignancy, severe dermatologic reactions, QT prolongation, hepatotoxicity, photosensitivity, and ophthalmologic toxicity, [see Warnings and Precautions (5)], advise women not to breastfeed during treatment with ZELBORAF and for 2 weeks after the final dose.
11 Description (11 DESCRIPTION)
ZELBORAF (vemurafenib) is a kinase inhibitor available as 240 mg tablets for oral use. Vemurafenib has the chemical name propane-1-sulfonic acid {3-[5-(4-chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl]- 2,4-difluoro-phenyl}-amide. It has the molecular formula C23H18ClF2N3O3S and a molecular weight of 489.9. Vemurafenib has the following chemical structure:
Vemurafenib is a white to off-white crystalline solid. It is practically insoluble in aqueous media.
Tablets of ZELBORAF are for oral administration. Each tablet contains 240 mg of vemurafenib.
The inactive ingredients of ZELBORAF are: Tablet core: hypromellose acetate succinate, croscarmellose sodium, colloidal silicon dioxide, magnesium stearate, and hydroxypropyl cellulose. Coating: pinkish white: poly (vinyl alcohol), titanium dioxide, polyethylene glycol 3350, talc, and iron oxide red.
8.4 Pediatric Use
The safety and effectiveness of ZELBORAF in pediatric patients have not been established. Vemurafenib was studied in 6 adolescent patients 15 to 17 years of age with unresectable or metastatic melanoma with BRAF V600 mutation. A maximum tolerated dose was not reached with doses up to vemurafenib 960 mg twice daily. No new safety signals were observed. Vemurafenib steady-state exposure in these 6 adolescent patients was generally similar to that in adults.
8.5 Geriatric Use
Clinical studies of ZELBORAF did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
5.11 Renal Failure
Renal failure, including acute interstitial nephritis and acute tubular necrosis, can occur with ZELBORAF. In Trial 1, in patients with metastatic melanoma, 26% of ZELBORAF-treated patients and 5% of dacarbazine-treated patients experienced Grade 1-2 creatinine elevations [greater than 1 and up to 3 times upper limit of normal (ULN)]; 1.2% of ZELBORAF-treated patients and 1.1% of dacarbazine-treated patients experienced Grade 3-4 creatinine elevations (greater than 3 times ULN).
In Trial 4, in patients with ECD, 86% (19/22) of patients experienced Grade 1/2 creatinine elevations and 9.1% (2/22) of patients experienced Grade 3 creatinine elevations.
Measure serum creatinine before initiation of ZELBORAF and periodically during treatment.
5.6 Hepatotoxicity
Liver injury leading to functional hepatic impairment, including coagulopathy or other organ dysfunction, can occur with ZELBORAF [see Adverse Reactions (6.1)]. Monitor transaminases, alkaline phosphatase, and bilirubin before initiation of treatment and monthly during treatment, or as clinically indicated. Manage laboratory abnormalities with dose reduction, treatment interruption, or treatment discontinuation [see Dosage and Administration (2.3)].
4 Contraindications (4 CONTRAINDICATIONS)
None.
5.5 Qt Prolongation (5.5 QT Prolongation)
Concentration-dependent QT prolongation occurred in an uncontrolled, open-label QT sub-study in previously treated patients with BRAF V600E mutation-positive metastatic melanoma [see Clinical Pharmacology (12.2)]. QT prolongation may lead to an increased risk of ventricular arrhythmias, including Torsade de Pointes.
Do not start treatment in patients with uncorrectable electrolyte abnormalities, QTc > 500 ms, or long QT syndrome, or in patients who are taking medicinal products known to prolong the QT interval. Prior to and following treatment initiation or after dose modification of ZELBORAF for QTc prolongation, evaluate ECG and electrolytes (including potassium, magnesium, and calcium) after 15 days, monthly during the first 3 months, and then every 3 months thereafter or more often as clinically indicated.
Withhold ZELBORAF in patients who develop QTc > 500 ms (Grade 3). Upon recovery to QTc ≤ 500 ms (Grade ≤ 2), restart at a reduced dose. Permanently discontinue ZELBORAF treatment if the QTc interval remains > 500 ms and increased > 60 ms from pre-treatment values after controlling cardiac risk factors for QT prolongation (e.g., electrolyte abnormalities, congestive heart failure, and bradyarrhythmias) [see Dosage and Administration (2.3)].
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following adverse reactions are discussed in greater detail in other sections of the label:
- New Primary Malignancies [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Dermatologic Reactions [see Warnings and Precautions (5.4)]
- QT Prolongation [see Warnings and Precautions (5.5)]
- Hepatotoxicity [see Warnings and Precautions (5.6)]
- Photosensitivity [see Warnings and Precautions (5.7)]
- Ophthalmologic Reactions [see Warnings and Precautions (5.8)]
- Radiation Sensitization and Radiation Recall [see Warnings and Precautions (5.10)]
- Renal Failure [see Warnings and Precautions (5.11)]
- Dupuytren's Contracture and Plantar Fascial Fibromatosis [see Warnings and Precautions (5.12)]
7 Drug Interactions (7 DRUG INTERACTIONS)
- Avoid concomitant administration of ZELBORAF with strong CYP3A4 inhibitors or inducers. (7.1)
- CYP1A2 Substrates: ZELBORAF can increase concentrations of CYP1A2 substrates. Avoid concomitant use of ZELBORAF with CYP1A2 substrates with a narrow therapeutic window. If coadministration cannot be avoided, monitor closely for toxicities and consider dose reduction of CYP1A2 substrates. (7.2).
2.2 Recommended Dose
The recommended dose of ZELBORAF is 960 mg (four 240 mg tablets) orally every 12 hours with or without a meal. A missed dose can be taken up to 4 hours prior to the next dose.
Treat patients with ZELBORAF until disease progression or unacceptable toxicity occurs.
Do not take an additional dose if vomiting occurs after ZELBORAF administration, but continue with the next scheduled dose.
Do not crush or chew the tablets.
5.7 Photosensitivity
Mild to severe photosensitivity can occur in patients treated with ZELBORAF [see Adverse Reactions (6.1)]. Advise patients to avoid sun exposure, wear protective clothing and use a broad spectrum UVA/UVB sunscreen and lip balm (SPF ≥ 30) when outdoors.
Institute dose modifications for intolerable Grade 2 or greater photosensitivity [see Dosage and Administration (2.2)].
8.7 Renal Impairment
No formal clinical study has been conducted to evaluate the effect of renal impairment on the pharmacokinetics of vemurafenib. No dose adjustment is recommended for patients with mild and moderate renal impairment based on a population pharmacokinetic analysis [see Clinical Pharmacology (12.3)]. The appropriate dose of ZELBORAF has not been established in patients with severe renal impairment.
12.3 Pharmacokinetics
The pharmacokinetics of vemurafenib were determined in patients with BRAF mutation-positive metastatic melanoma following 15 days of 960 mg twice daily with dosing approximately 12 hours apart. The population pharmacokinetic analysis pooled data from 458 patients. At steady-state, vemurafenib exhibits linear pharmacokinetics within the 240 mg to 960 mg dose range.
8.6 Hepatic Impairment
No formal clinical study has been conducted to evaluate the effect of hepatic impairment on the pharmacokinetics of vemurafenib. No dose adjustment is recommended for patients with mild and moderate hepatic impairment based on a population pharmacokinetic analysis [see Clinical Pharmacology (12.3)]. The appropriate dose of ZELBORAF has not been established in patients with severe hepatic impairment.
1 Indications and Usage (1 INDICATIONS AND USAGE)
- ZELBORAF ® is a kinase inhibitor indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test. (1.1, 2.1)
- ZELBORAF® is indicated for the treatment of patients with Erdheim- Chester Disease with BRAF V600 mutation. (1.2, 2.1)
Limitation of Use: ZELBORAF is not indicated for treatment of patients with wild-type BRAF melanoma (2.1, 5.2)
12.1 Mechanism of Action
Vemurafenib is a low molecular weight, orally available inhibitor of some mutated forms of BRAF serine- threonine kinase, including BRAF V600E. Vemurafenib also inhibits other kinases in vitro such as CRAF, ARAF, wild-type BRAF, SRMS, ACK1, MAP4K5, and FGR at similar concentrations. Some mutations in the BRAF gene including V600E result in constitutively activated BRAF proteins, which can cause cell proliferation in the absence of growth factors that would normally be required for proliferation. Vemurafenib has anti-tumor effects in cellular and animal models of melanomas with mutated BRAF V600E.
5.9 Embryo Fetal Toxicity (5.9 Embryo-Fetal Toxicity)
Based on its mechanism of action, ZELBORAF can cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ZELBORAF and for 2 weeks after the final dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
7.3 Concurrent Ipilimumab
Increases in transaminases and bilirubin occurred in a majority of patients who received concurrent ipilimumab and ZELBORAF [see Warnings and Precautions Section 5.6].
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- New Primary Cutaneous Malignancies: Perform dermatologic evaluations prior to initiation of therapy, every 2 months while on therapy, and for up to 6 months following discontinuation of ZELBORAF. Manage with excision and continue treatment without dose adjustment. (5.1)
- New Non-Cutaneous Squamous Cell Carcinoma: Evaluate for symptoms or clinical signs of new non-cutaneous SCC before initiation of treatment and periodically during treatment. (5.1)
- Other Malignancies: Monitor patients receiving ZELBORAF closely for signs or symptoms of other malignancies (5.1).
- Tumor Promotion in BRAF Wild-Type Melanoma: Increased cell proliferation can occur with BRAF inhibitors (5.2).
- Serious Hypersensitivity Reactions including anaphylaxis and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS Syndrome): Discontinue ZELBORAF for severe hypersensitivity reactions. (5.3)
- Severe Dermatologic Reactions, including Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: Discontinue ZELBORAF for severe dermatologic reactions. (5.4)
- QT Prolongation: Monitor ECG and electrolytes before and during treatment. Withhold ZELBORAF for QTc of 500 ms or greater. Correct electrolyte abnormalities and control for cardiac risk factors for QT prolongation. (5.5)
- Hepatotoxicity: Measure liver enzymes and bilirubin before initiating ZELBORAF and monitor monthly during treatment. (5.6)
- Photosensitivity: Advise patients to avoid sun exposure. (5.7)
- Serious Ophthalmologic Reactions: Monitor for signs and symptoms of uveitis. (5.8)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of the potential risk to the fetus and to use effective contraception. (5.9, 8.1, 8.3)
- Radiation Sensitization and Radiation Recall: Severe cases have been reported. (5.10).
- Renal Failure: Measure serum creatinine before initiating ZELBORAF and monitor periodically during treatment (5.11).
- Dupuytren's Contracture and plantar fascial fibromatosis: Events should be managed with dose reduction, treatment interruption, or treatment discontinuation. (5.12).
5.4 Dermatologic Reactions
Severe dermatologic reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis, can occur in patients receiving ZELBORAF. Permanently discontinue ZELBORAF in patients who experience a severe dermatologic reaction [see Adverse Reactions (6.1)].
1.2 Erdheim Chester Disease (1.2 Erdheim-Chester Disease)
ZELBORAF® is indicated for the treatment of patients with Erdheim-Chester Disease (ECD) with BRAF V600 mutation.
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Tablet: 240 mg.
5.8 Ophthalmologic Reactions
Uveitis, blurry vision, and photophobia can occur in patients treated with ZELBORAF. In Trial 1, uveitis, including iritis, occurred in 2.1% (7/336) of patients receiving ZELBORAF compared to no patients in the dacarbazine arm. Treatment with steroid and mydriatic ophthalmic drops may be required to manage uveitis. Monitor patients for signs and symptoms of uveitis.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ZELBORAF. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Neoplasms benign, malignant and unspecified (incl. cysts and polyps): Progression of pre-existing chronic myelomonocytic leukemia with NRAS mutation [see Warnings and Precautions (5.1)].
Skin and subcutaneous tissue disorders: Drug reaction with eosinophilia and systemic symptoms (DRESS syndrome) [see Warnings and Precautions (5.3)].
Blood and lymphatic systems disorder: Neutropenia
Injury, poisoning and procedural complications: Radiation sensitization and recall [see Warnings and Precautions (5.10)].
Gastrointestinal disorders: Pancreatitis
Renal and urinary disorders: Acute interstitial nephritis, acute tubular necrosis [see Warnings and Precautions (5.11)].
Musculoskeletal and connective tissue disorders: Dupuytren's contracture and plantar fascial fibromatosis [see Warnings and Precautions (5.12)].
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
- Lactation: Do not breastfeed while taking ZELBORAF. (8.2)
5.3 Hypersensitivity Reactions
Anaphylaxis and other serious hypersensitivity reactions can occur during treatment and upon re-initiation of treatment with ZELBORAF. Severe hypersensitivity reactions included generalized rash and erythema, hypotension, and drug reaction with eosinophilia and systemic symptoms (DRESS syndrome). Permanently discontinue ZELBORAF in patients who experience a severe hypersensitivity reaction [see Adverse Reactions (6.2)].
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
Unresectable or Metastatic Melanoma with BRAF V600E Mutation This section describes adverse drug reactions (ADRs) identified from analyses of Trial 1 and Trial 2 [see Clinical Studies (14)]. Trial 1 randomized (1:1) 675 treatment-naive patients with unresectable or metastatic melanoma to receive ZELBORAF 960 mg orally twice daily or dacarbazine 1000 mg/m2 intravenously every 3 weeks. In Trial 2, 132 patients with metastatic melanoma and failure of at least one prior systemic therapy received treatment with ZELBORAF 960 mg orally twice daily.
Table 1 presents adverse reactions reported in at least 10% of unresectable or metastatic melanoma patients treated with ZELBORAF. The most common adverse reactions of any grade (≥ 30% in either study) in ZELBORAF-treated patients were arthralgia, rash, alopecia, fatigue, photosensitivity reaction, nausea, pruritus, and skin papilloma. The most common (≥ 5%) Grade 3 adverse reactions were cuSCC and rash. The incidence of Grade 4 adverse reactions was ≤ 4% in both studies.
The incidence of adverse events resulting in permanent discontinuation of study medication in Trial 1 was 7% for the ZELBORAF arm and 4% for the dacarbazine arm. In Trial 2, the incidence of adverse events resulting in permanent discontinuation of study medication was 3% in ZELBORAF-treated patients. The median duration of study treatment was 4.2 months for ZELBORAF and 0.8 months for dacarbazine in Trial 1, and 5.7 months for ZELBORAF in Trial 2.
| ADRs | Trial 1: Treatment-Naïve Patients | Trial 2: Patients with Failure of at Least One Prior Systemic Therapy | ||||
|---|---|---|---|---|---|---|
| ZELBORAF n=336 |
Dacarbazine n=287 |
ZELBORAF n=132 |
||||
| All Grades (%) |
Grade 3 Grade 4 adverse reactions limited to gamma-glutamyltransferase increased (< 1% in Trial 1 and 4% in Trial 2).
(%) |
All Grades (%) |
Grade 3 (%) |
All Grades (%) | Grade 3 (%) | |
| Skin and subcutaneous tissue disorders | ||||||
| Rash | 37 | 8 | 2 | 0 | 52 | 7 |
| Photosensitivity reaction | 33 | 3 | 4 | 0 | 49 | 3 |
| Alopecia | 45 | < 1 | 2 | 0 | 36 | 0 |
| Pruritus | 23 | 1 | 1 | 0 | 30 | 2 |
| Hyperkeratosis | 24 | 1 | < 1 | 0 | 28 | 0 |
| Rash maculo-papular | 9 | 2 | < 1 | 0 | 21 | 6 |
| Actinic keratosis | 8 | 0 | 3 | 0 | 17 | 0 |
| Dry skin | 19 | 0 | 1 | 0 | 16 | 0 |
| Rash papular | 5 | < 1 | 0 | 0 | 13 | 0 |
| Erythema | 14 | 0 | 2 | 0 | 8 | 0 |
| Musculoskeletal and connective tissue disorders | ||||||
| Arthralgia | 53 | 4 | 3 | < 1 | 67 | 8 |
| Myalgia | 13 | < 1 | 1 | 0 | 24 | < 1 |
| Pain in extremity | 18 | < 1 | 6 | 2 | 9 | 0 |
| Musculoskeletal pain | 8 | 0 | 4 | < 1 | 11 | 0 |
| Back pain | 8 | < 1 | 5 | < 1 | 11 | < 1 |
| General disorders and administration site conditions | ||||||
| Fatigue | 38 | 2 | 33 | 2 | 54 | 4 |
| Edema peripheral | 17 | < 1 | 5 | 0 | 23 | 0 |
| Pyrexia | 19 | < 1 | 9 | < 1 | 17 | 2 |
| Asthenia | 11 | < 1 | 9 | < 1 | 2 | 0 |
| Gastrointestinal disorders | ||||||
| Nausea | 35 | 2 | 43 | 2 | 37 | 2 |
| Diarrhea | 28 | < 1 | 13 | < 1 | 29 | < 1 |
| Vomiting | 18 | 1 | 26 | 1 | 26 | 2 |
| Constipation | 12 | < 1 | 24 | 0 | 16 | 0 |
| Nervous system disorders | ||||||
| Headache | 23 | < 1 | 10 | 0 | 27 | 0 |
| Dysgeusia | 14 | 0 | 3 | 0 | 11 | 0 |
| Neoplasms benign, malignant and unspecified (includes cysts and polyps) | ||||||
| Skin papilloma | 21 | < 1 | 0 | 0 | 30 | 0 |
| Cutaneous SCC Includes both squamous cell carcinoma of the skin and keratoacanthoma.
Cases of cutaneous squamous cell carcinoma were required to be reported as Grade 3 per protocol.
|
24 | 22 | < 1 | < 1 | 24 | 24 |
| Seborrheic keratosis | 10 | < 1 | 1 | 0 | 14 | 0 |
| Investigations | ||||||
| Gamma-glutamyltransferase increased | 5 | 3 | 1 | 0 | 15 | 6 |
| Metabolism and nutrition disorders | ||||||
| Decreased appetite | 18 | 0 | 8 | < 1 | 21 | 0 |
| Respiratory, thoracic and mediastinal disorders | ||||||
| Cough | 8 | 0 | 7 | 0 | 12 | 0 |
| Injury, poisoning and procedural complications | ||||||
| Sunburn | 10 | 0 | 0 | 0 | 14 | 0 |
Clinically relevant adverse reactions reported in < 10% of unresectable or metastatic melanoma patients treated with ZELBORAF in the Phase 2 and Phase 3 studies include:
Skin and subcutaneous tissue disorders: palmar-plantar erythrodysesthesia syndrome, keratosis pilaris, panniculitis, erythema nodosum, Stevens-Johnson syndrome, toxic epidermal necrolysis
Musculoskeletal and connective tissue disorders: arthritis, Dupuytren's contracture
Nervous system disorders: neuropathy peripheral, VIIth nerve paralysis
Neoplasms benign, malignant and unspecified (includes cysts and polyps): basal cell carcinoma, oropharyngeal squamous cell carcinoma
Infections and infestations: folliculitis
Eye disorders: retinal vein occlusion
Vascular disorders: vasculitis
Cardiac disorders: atrial fibrillation
Table 2 shows the incidence of worsening liver laboratory abnormalities in Trial 1 summarized as the proportion of patients who experienced a shift from baseline to Grade 3 or 4.
| Parameter | Change From Baseline to Grade 3/4 | |
|---|---|---|
| ZELBORAF (%) | Dacarbazine (%) | |
| GGT | 11.5 | 8.6 |
| AST | 0.9 | 0.4 |
| ALT | 2.8 | 1.9 |
| Alkaline phosphatase | 2.9 | 0.4 |
| Bilirubin | 1.9 | 0 |
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Healthcare providers should advise patients of the potential benefits and risks of ZELBORAF and instruct their patients to read the Medication Guide before starting ZELBORAF therapy. Inform patients of the following:
- Evidence of BRAF V600E mutation in the tumor specimen with an FDA approved test is necessary to identify patients with melanoma for whom treatment with ZELBORAF is indicated [see Dosage and Administration (2.1)].
- ZELBORAF increases the risk of developing new primary cutaneous malignancies. Advise patients of the importance of contacting their healthcare provider immediately for any changes in their skin [see Warnings and Precautions (5.1)].
- Anaphylaxis and other serious hypersensitivity reactions can occur during treatment and upon re- initiation of treatment with ZELBORAF. Advise patients to stop taking ZELBORAF and to seek immediate medical attention for symptoms of anaphylaxis or hypersensitivity [see Warnings and Precautions (5.3)].
- Severe dermatologic reactions can occur in patients receiving ZELBORAF. Advise patients to stop taking ZELBORAF and to contact their health-care provider for severe dermatologic reactions [see Warnings and Precautions (5.4)].
- ZELBORAF can prolong QT interval, which may result in ventricular arrhythmias. Advise patients of the importance of monitoring of their electrolytes and the electrical activity of their heart (via an ECG) during ZELBORAF treatment [see Warnings and Precautions (5.5)].
- Liver injury leading to functional hepatic impairment, including coagulopathy or other organ dysfunction, can occur with ZELBORAF. Advise patients of the importance of laboratory monitoring of their liver during ZELBORAF treatment and to contact their health-care provider for relevant symptoms [see Warnings and Precautions (5.6)].
- ZELBORAF can cause mild to severe photosensitivity. Advise patients to avoid sun exposure, wear protective clothing, and use a broad spectrum UVA/UVB sunscreen and lip balm (SPF ≥ 30) when outdoors to help protect against sunburn [see Warnings and Precautions (5.7)].
- Ophthalmologic reactions can occur in patients treated with ZELBORAF. Advise patients to contact their health-care provider immediately for ophthalmologic symptoms [see Warnings and Precautions (5.8)].
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
ZELBORAF (vemurafenib) is supplied as 240 mg film-coated tablets with VEM debossed on one side. The following packaging configurations are available:
NDC 50242-090-01 single bottle of 120 count
NDC 50242-090-02 single bottle of 112 count
1.1 Unresectable Or Metastatic Melanoma (1.1 Unresectable or Metastatic Melanoma)
ZELBORAF ® is indicated for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E mutation as detected by an FDA-approved test.
13.2 Animal Toxicology And/or Pharmacology (13.2 Animal Toxicology and/or Pharmacology)
Consistent with the increased incidence of cutaneous squamous cell carcinomas in patients treated with vemurafenib, the treatment of mice implanted with human cuSCC cells with vemurafenib caused a dose- dependent acceleration of the growth of the implanted tumors.
7.4 Effect of Vemurafenib On P Gp Substrates (7.4 Effect of Vemurafenib on P-gp Substrates)
Coadministration of ZELBORAF with digoxin, a sensitive P-glycoprotein (P-gp) substrate, increased digoxin systemic exposure by 1.8-fold. Avoid concurrent use of P-gp substrates known to have narrow therapeutic indices. If use of these medications is unavoidable, consider dose reduction of P-gp substrates with narrow therapeutic indices.
5.2 Tumor Promotion in Braf Wild Type Melanoma (5.2 Tumor Promotion in BRAF Wild-Type Melanoma)
In vitro experiments have demonstrated paradoxical activation of MAP-kinase signaling and increased cell proliferation in BRAF wild-type cells that are exposed to BRAF inhibitors. Confirm evidence of BRAF V600E mutation in tumor specimens prior to initiation of ZELBORAF [see Indications and Usage (1) and Dosage and Administration (2.1)].
7.2 Effect of Vemurafenib On Cyp1a2 Substrates (7.2 Effect of Vemurafenib on CYP1A2 Substrates)
Coadministration of ZELBORAF with tizanidine, a sensitive CYP1A2 substrate, increased tizanidine systemic exposure by 4.7-fold. Avoid concomitant use of ZELBORAF with drugs having a narrow therapeutic window that are predominantly metabolized by CYP1A2 [see Clinical Pharmacology (12.3)]. If coadministration cannot be avoided, monitor closely for toxicities and consider a dose reduction of concomitant CYP1A2 substrates.
2.1 Patient Selection for Treatment of Melanoma
Confirm the presence of BRAF V600E mutation in melanoma tumor specimens prior to initiation of treatment with ZELBORAF [see Warnings and Precautions (5.2)]. Information on FDA-approved tests for the detection of BRAF V600 mutations in melanoma is available at http://www.fda.gov/CompanionDiagnostics.
2.4 Dose Modification for Strong Cyp3a4 Inducers (2.4 Dose Modification for Strong CYP3A4 Inducers)
Avoid concomitant use of strong CYP3A4 inducers during treatment with ZELBORAF [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]. If concomitant use of a strong CYP3A4 inducer is unavoidable, increase the dose of ZELBORAF by 240 mg (one tablet) as tolerated. After discontinuation of a strong CYP3A4 inducer for two weeks, resume the ZELBORAF dose that was taken prior to initiating the strong CYP3A4 inducer.
5.10 Radiation Sensitization and Radiation Recall
Radiation sensitization and recall, in some cases severe, involving cutaneous and visceral organs have been reported in patients treated with radiation prior to, during, or subsequent to vemurafenib treatment. Fatal cases have been reported in patients with visceral organ involvement. [see Adverse Reactions (6.2)].
Monitor patients closely when vemurafenib is administered concomitantly or sequentially with radiation treatment.
Principal Display Panel 240 Mg Tablet Bottle Carton (PRINCIPAL DISPLAY PANEL - 240 mg Tablet Bottle Carton)
NDC 50242-090-02
Zelboraf®
(vemurafenib)
tablets
240 mg
Do not crush or chew tablet.
Rx only
Attention Pharmacist: Dispense the
accompanying Medication Guide to
each patient.
112 tablets
Genentech | Daiichi Sankyo, Inc.
10210025
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
There have been no formal studies conducted assessing the carcinogenic potential of vemurafenib. ZELBORAF increased the development of cutaneous squamous cell carcinomas in patients in clinical trials.
Vemurafenib did not cause genetic damage when tested in in vitro assays (bacterial mutation [AMES Assay], human lymphocyte chromosome aberration) or in the in vivo rat bone marrow micronucleus test.
No specific studies with vemurafenib have been conducted in animals to evaluate the effect on fertility; nevertheless, no histopathological findings were noted in reproductive organs in males and females in repeat-dose toxicology studies in rats at doses up to 450 mg/kg/day (approximately 0.6 and 1.6 times the human exposure based on AUC in males and females, respectively) and dogs at doses up to 450 mg/kg/day (approximately 0.3 times the human clinical exposure based on AUC in both males and females, respectively).
5.12 Dupuytren's Contracture and Plantar Fascial Fibromatosis
Dupuytren's contracture and plantar fascial fibromatosis have been reported with ZELBORAF. The majority of cases were mild to moderate, but severe, disabling cases of Dupuytren's contracture have also been reported [see Dosage and Administration (2.3), Adverse Reactions (6.1, 6.2)].
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:50:57.758085 · Updated: 2026-03-14T22:38:58.661986