These Highlights Do Not Include All The Information Needed To Use Columvi®
3516e753-f064-4d30-9bd5-e3f2e143f75d
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
COLUMVI is indicated for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma, not otherwise specified (DLBCL, NOS) or large B-cell lymphoma (LBCL) arising from follicular lymphoma, after two or more lines of systemic therapy. This indication is approved under accelerated approval based on response rate and durability of response [see Clinical Studies (14.1) ] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Dosage and Administration
Pretreat with a single 1,000 mg dose of obinutuzumab intravenously 7 days before initiation of COLUMVI (Cycle 1 Day 1). ( 2.2 ) Administer premedications as recommended. ( 2.3 ) Administer only as an intravenous infusion. ( 2.1 ) Recommended dosage ( 2.2 ): Treatment Cycle Cycle = 21 days Day Dose of COLUMVI Day 1 Obinutuzumab 1,000 mg Cycle 1 Day 8 Step-up dose 1 2.5 mg Day 15 Step-up dose 2 10 mg Cycle 2 to 12 Day 1 30 mg Administer in a facility equipped to monitor and manage CRS. ( 2.1 , 2.2 ) Patients should be hospitalized for the 2.5 mg step-up dose and for subsequent infusions as recommended. ( 2.1 , 2.2 ) See Full Prescribing Information for instructions on preparation and administration. ( 2.5 , 2.6 , 2.7 )
Contraindications
None.
Warnings and Precautions
Neurologic Toxicity : Can cause serious neurologic toxicity, including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS). Monitor for neurologic toxicity; withhold or permanently discontinue based on severity. ( 5.2 ) Serious Infections : Can cause serious or fatal infections. Monitor patients for signs and symptoms of infection and treat appropriately. ( 5.3 ) Tumor Flare : Can cause serious tumor flare reactions. Monitor patients at risk for complications of tumor flare. ( 5.4 ) Embryo-Fetal Toxicity : May cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception. ( 5.5 , 8.1 , 8.3 )
Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling: Cytokine Release Syndrome [see Warnings and Precautions (5.1) ] Neurologic Toxicity [see Warnings and Precautions (5.2) ] Serious Infections [see Warnings and Precautions (5.3) ] Tumor Flare [see Warnings and Precautions (5.4) ]
Drug Interactions
For certain CYP substrates where minimal concentration changes may lead to serious adverse reactions, monitor for toxicities or drug concentrations of such CYP substrates when coadministered with COLUMVI. Glofitamab-gxbm causes the release of cytokines [see Clinical Pharmacology (12.2) ] that may suppress the activity of CYP enzymes, resulting in increased exposure of CYP substrates. Increased exposure of CYP substrates is more likely to occur after the first dose of COLUMVI on Cycle 1 Day 8 and up to 14 days after the first 30 mg dose on Cycle 2 Day 1 and during and after CRS [see Warnings and Precautions (5.1) ] .
How Supplied
COLUMVI (glofitamab-gxbm) injection is a sterile, preservative-free, colorless, clear solution for intravenous infusion. COLUMVI is supplied as: Carton Contents NDC One 2.5 mg/2.5 mL (1 mg/mL) single-dose vial NDC 50242-125-01 One 10 mg/10 mL (1 mg/mL) single-dose vial NDC 50242-127-01
Storage and Handling
COLUMVI (glofitamab-gxbm) injection is a sterile, preservative-free, colorless, clear solution for intravenous infusion. COLUMVI is supplied as: Carton Contents NDC One 2.5 mg/2.5 mL (1 mg/mL) single-dose vial NDC 50242-125-01 One 10 mg/10 mL (1 mg/mL) single-dose vial NDC 50242-127-01
Description
Cytokine Release Syndrome (CRS), including serious or fatal reactions, can occur in patients receiving COLUMVI. Premedicate before each dose, and initiate treatment with the COLUMVI step-up dosing schedule to reduce the risk of CRS. Withhold COLUMVI until CRS resolves or permanently discontinue based on severity [see Dosage and Administration (2.1 , 2.2 , 2.3 , and 2.4) and Warnings and Precautions (5.1) ] .
Medication Information
Warnings and Precautions
Neurologic Toxicity : Can cause serious neurologic toxicity, including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS). Monitor for neurologic toxicity; withhold or permanently discontinue based on severity. ( 5.2 ) Serious Infections : Can cause serious or fatal infections. Monitor patients for signs and symptoms of infection and treat appropriately. ( 5.3 ) Tumor Flare : Can cause serious tumor flare reactions. Monitor patients at risk for complications of tumor flare. ( 5.4 ) Embryo-Fetal Toxicity : May cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception. ( 5.5 , 8.1 , 8.3 )
Indications and Usage
COLUMVI is indicated for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma, not otherwise specified (DLBCL, NOS) or large B-cell lymphoma (LBCL) arising from follicular lymphoma, after two or more lines of systemic therapy. This indication is approved under accelerated approval based on response rate and durability of response [see Clinical Studies (14.1) ] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Dosage and Administration
Pretreat with a single 1,000 mg dose of obinutuzumab intravenously 7 days before initiation of COLUMVI (Cycle 1 Day 1). ( 2.2 ) Administer premedications as recommended. ( 2.3 ) Administer only as an intravenous infusion. ( 2.1 ) Recommended dosage ( 2.2 ): Treatment Cycle Cycle = 21 days Day Dose of COLUMVI Day 1 Obinutuzumab 1,000 mg Cycle 1 Day 8 Step-up dose 1 2.5 mg Day 15 Step-up dose 2 10 mg Cycle 2 to 12 Day 1 30 mg Administer in a facility equipped to monitor and manage CRS. ( 2.1 , 2.2 ) Patients should be hospitalized for the 2.5 mg step-up dose and for subsequent infusions as recommended. ( 2.1 , 2.2 ) See Full Prescribing Information for instructions on preparation and administration. ( 2.5 , 2.6 , 2.7 )
Contraindications
None.
Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling: Cytokine Release Syndrome [see Warnings and Precautions (5.1) ] Neurologic Toxicity [see Warnings and Precautions (5.2) ] Serious Infections [see Warnings and Precautions (5.3) ] Tumor Flare [see Warnings and Precautions (5.4) ]
Drug Interactions
For certain CYP substrates where minimal concentration changes may lead to serious adverse reactions, monitor for toxicities or drug concentrations of such CYP substrates when coadministered with COLUMVI. Glofitamab-gxbm causes the release of cytokines [see Clinical Pharmacology (12.2) ] that may suppress the activity of CYP enzymes, resulting in increased exposure of CYP substrates. Increased exposure of CYP substrates is more likely to occur after the first dose of COLUMVI on Cycle 1 Day 8 and up to 14 days after the first 30 mg dose on Cycle 2 Day 1 and during and after CRS [see Warnings and Precautions (5.1) ] .
Storage and Handling
COLUMVI (glofitamab-gxbm) injection is a sterile, preservative-free, colorless, clear solution for intravenous infusion. COLUMVI is supplied as: Carton Contents NDC One 2.5 mg/2.5 mL (1 mg/mL) single-dose vial NDC 50242-125-01 One 10 mg/10 mL (1 mg/mL) single-dose vial NDC 50242-127-01
How Supplied
COLUMVI (glofitamab-gxbm) injection is a sterile, preservative-free, colorless, clear solution for intravenous infusion. COLUMVI is supplied as: Carton Contents NDC One 2.5 mg/2.5 mL (1 mg/mL) single-dose vial NDC 50242-125-01 One 10 mg/10 mL (1 mg/mL) single-dose vial NDC 50242-127-01
Description
Cytokine Release Syndrome (CRS), including serious or fatal reactions, can occur in patients receiving COLUMVI. Premedicate before each dose, and initiate treatment with the COLUMVI step-up dosing schedule to reduce the risk of CRS. Withhold COLUMVI until CRS resolves or permanently discontinue based on severity [see Dosage and Administration (2.1 , 2.2 , 2.3 , and 2.4) and Warnings and Precautions (5.1) ] .
Section 42229-5
Pretreatment with Obinutuzumab
Pretreat all patients with a single 1,000 mg dose of obinutuzumab administered as an intravenous infusion on Cycle 1 Day 1, 7 days prior to initiation of COLUMVI (see Table 1) to deplete the circulating and lymphoid tissue B cells.
Obinutuzumab should be administered as an intravenous infusion at 50 mg/hour. The rate of infusion can be escalated in 50 mg/hour increments every 30 minutes to a maximum of 400 mg/hour. Refer to the obinutuzumab prescribing information for complete dosing information.
Section 42231-1
| MEDICATION GUIDE COLUMVI® (ko-loom-vee) (glofitamab-gxbm) injection, for intravenous infusion |
||
|---|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 6/2025 | |
|
What is the most important information I should know about COLUMVI? COLUMVI can cause Cytokine Release Syndrome (CRS), a serious side effect that is common during treatment with COLUMVI, and can also be serious and lead to death.
|
||
|
|
|
|
||
|
What is COLUMVI?
COLUMVI is a prescription medicine used to treat adults with certain types of diffuse large B-cell lymphoma (DLBCL) or large B-cell lymphoma (LBCL) that has come back (relapsed) or that did not respond to previous treatment (refractory), and who have received 2 or more prior treatments for their cancer. It is not known if COLUMVI is safe and effective in children. |
||
Before receiving COLUMVI, tell your healthcare provider about all of your medical conditions, including if you:
|
||
|
How will I receive COLUMVI?
|
||
|
What should I avoid while receiving COLUMVI? Do not drive, operate heavy machinery, or do other dangerous activities if you develop dizziness, confusion, shaking (tremors), sleepiness, or any other symptoms that impair consciousness until your signs and symptoms go away. These may be signs and symptoms of neurologic problems. See " What are the possible side effects of COLUMVI?" for more information about signs and symptoms of neurologic problems. |
||
|
What are the possible side effects of COLUMVI? COLUMVI may cause serious side effects, including:
|
||
|
|
|
|
||
| Your healthcare provider may temporarily stop or completely stop treatment with COLUMVI if you develop certain side effects. The most common side effects of COLUMVI include: CRS, muscle and bone pain, rash, and tiredness. The most common severe abnormal lab test results with COLUMVI include: decreased white blood cells, decreased phosphate (an electrolyte), increased uric acid levels, and decreased fibrinogen (a protein that helps with blood clotting). These are not all the possible side effects of COLUMVI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
|
General information about the safe and effective use of COLUMVI.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your healthcare provider or pharmacist for information about COLUMVI that is written for health professionals. |
||
|
What are the ingredients in COLUMVI? Active ingredient: glofitamab-gxbm Inactive ingredients: histidine, histidine hydrochloride monohydrate, methionine, polysorbate 20, sucrose, and Water for injection. Manufactured by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 U.S. License No.: 1048 For more information, go to www.COLUMVI.com or call 1-877-436-3683. |
Section 44425-7
Store refrigerated at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not freeze. Do not shake.
11 Description
Glofitamab-gxbm is a bispecific CD20-directed CD3 T-cell engager. It is a recombinant humanized anti-CD20 anti-CD3ɛ bispecific immunoglobulin G1 (IgG1) monoclonal antibody produced in Chinese hamster ovary (CHO) cells. Glofitamab-gxbm has an approximate molecular weight of 197 kDa.
COLUMVI (glofitamab-gxbm) injection is a sterile, preservative-free, colorless, clear solution supplied in single-dose vials for intravenous infusion.
COLUMVI is supplied in 2.5 mg/2.5 mL and 10 mg/10 mL single-dose vials at a concentration of 1 mg/mL. Each mL of solution contains 1 mg glofitamab-gxbm, histidine (0.63 mg), histidine hydrochloride monohydrate (3.34 mg), methionine (1.49 mg), polysorbate 20 (0.5 mg), sucrose (82.15 mg), and Water for Injection, USP, at pH 5.5.
5.4 Tumor Flare
COLUMVI can cause serious tumor flare [see Adverse Reactions (6.1)]. Manifestations include localized pain and swelling at the sites of the lymphoma lesions and/or dyspnea from new pleural effusions.
Tumor flare was reported in 12% of patients who received COLUMVI, including Grade 2 tumor flare in 4.8% of patients and Grade 3 tumor flare in 2.8%. Recurrent tumor flare occurred in two (12%) of the affected patients. Most tumor flare events occurred during Cycle 1, with a median time to first onset of 2 days (range: 1 to 16 days) after the first dose of COLUMVI. The median duration was 3.5 days (range: 1 to 35 days).
Patients with bulky tumors or disease located in close proximity to airways or a vital organ should be monitored closely during initial therapy. Monitor for signs and symptoms of compression or obstruction due to mass effect secondary to tumor flare, and institute appropriate treatment. Withhold COLUMVI until tumor flare resolves [see Dosage and Administration (2.4)].
8.4 Pediatric Use
The safety and efficacy of COLUMVI in pediatric patients have not been established.
8.5 Geriatric Use
Of the 145 patients with relapsed or refractory LBCL who received COLUMVI in study NP30179, 55% were 65 years of age or older, and 23% were 75 years of age or older. There was a higher rate of fatal adverse reactions, primarily from COVID-19, in patients 65 years of age or older compared to younger patients [see Adverse Reactions (6.1)]. No overall differences in efficacy were observed between patients 65 years of age or older and younger patients.
12.6 Immunogenicity
The observed incidence of antidrug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the study described below with the incidence of ADA in other studies, including those of glofitamab-gxbm.
During treatment in Study NP30179 (up to 9 months) [see Clinical Studies (14.1)], using an enzyme-linked immunosorbent assay (ELISA), the incidence of anti-glofitamab antibody formation was 1.1% (5/448) in patients treated with COLUMVI. Because of the low occurrence of ADAs, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of glofitamab-gxbm is unknown.
4 Contraindications
None.
6 Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Cytokine Release Syndrome [see Warnings and Precautions (5.1)]
- Neurologic Toxicity [see Warnings and Precautions (5.2)]
- Serious Infections [see Warnings and Precautions (5.3)]
- Tumor Flare [see Warnings and Precautions (5.4)]
7 Drug Interactions
For certain CYP substrates where minimal concentration changes may lead to serious adverse reactions, monitor for toxicities or drug concentrations of such CYP substrates when coadministered with COLUMVI.
Glofitamab-gxbm causes the release of cytokines [see Clinical Pharmacology (12.2)] that may suppress the activity of CYP enzymes, resulting in increased exposure of CYP substrates. Increased exposure of CYP substrates is more likely to occur after the first dose of COLUMVI on Cycle 1 Day 8 and up to 14 days after the first 30 mg dose on Cycle 2 Day 1 and during and after CRS [see Warnings and Precautions (5.1)].
12.3 Pharmacokinetics
The pharmacokinetics of glofitamab-gxbm was determined following pretreatment with a single dose of obinutuzumab of 1,000 mg and the pharmacokinetic parameters are presented as geometric mean (CV%) unless otherwise specified. Glofitamab-gxbm exposure increased dose-proportionally over the dose range from 0.005 to 30 mg (0.000167 to 1 time the recommended treatment dosage). Glofitamab-gxbm exposure parameters are summarized in Table 11 for the approved recommended dosage of COLUMVI.
| AUCtau (day∙mcg/mL) | Cmax (mcg/mL) | Ctrough (mcg/mL) | |
|---|---|---|---|
| Data presented as geometric mean (CV%). AUCtau = area under the concentration-time curve over one 21-day cycle; Cmax = maximum glofitamab-gxbm concentration; Ctrough = glofitamab-gxbm concentration prior to next dose; CV = geometric coefficient of variation. | |||
| First full 30 mg dose | 44.5 (55%) | 9.41 (27%) | 0.52 (83%) |
| Steady state Steady state values are approximated at Cycle 6 (week 18). 30 mg dose |
48.6 (33%) | 9.44 (26%) | 0.59 (67%) |
5.3 Serious Infections
COLUMVI can cause serious or fatal infections [see Adverse Reactions (6.1)].
Serious infections were reported in 16% of patients, including Grade 3 or 4 infections in 10%, and fatal infections in 4.8% of patients. Grade 3 or higher infections reported in ≥ 2% of patients were COVID-19 infection (6%), including COVID-19 pneumonia, and sepsis (4.1%). Febrile neutropenia occurred in 3.4% of patients.
COLUMVI should not be administered to patients with an active infection. Administer antimicrobial prophylaxis according to guidelines. Monitor patients before and during COLUMVI treatment for infection and treat appropriately. Withhold or consider permanent discontinuation of COLUMVI based on severity [see Dosage and Administration (2.4)].
1 Indications and Usage
COLUMVI is indicated for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma, not otherwise specified (DLBCL, NOS) or large B-cell lymphoma (LBCL) arising from follicular lymphoma, after two or more lines of systemic therapy.
This indication is approved under accelerated approval based on response rate and durability of response [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
5.2 Neurologic Toxicity
COLUMVI can cause serious and fatal neurologic toxicity, including Immune Effector Cell-Associated Neurotoxicity (ICANS) [see Adverse Reactions (6.1)].
Among 145 patients who received COLUMVI, the most frequent neurologic toxicities of any grade were headache (10%), peripheral neuropathy (8%), dizziness or vertigo (7%), and mental status changes (4.8%, including confusional state, cognitive disorder, disorientation, somnolence, and delirium). Grade 3 or higher neurologic adverse reactions occurred in 2.1% of patients and included somnolence, delirium, and myelitis. Cases of ICANS of any grade occurred in 4.8% of patients.
Coadministration of COLUMVI with other products that cause dizziness or mental status changes may increase the risk of neurologic toxicity. Optimize concomitant medications and hydration to avoid dizziness or mental status changes. Institute fall precautions as appropriate.
Monitor patients for signs and symptoms of neurologic toxicity, evaluate, and provide supportive therapy; withhold or permanently discontinue COLUMVI based on severity [see Dosage and Administration (2.4)].
Evaluate patients who experience neurologic toxicity such as tremors, dizziness, or adverse reactions that may impair cognition or consciousness promptly, including potential neurology evaluation. Advise affected patients to refrain from driving and/or engaging in hazardous occupations or activities, such as operating heavy or potentially dangerous machinery, until the neurologic toxicity fully resolves.
12.1 Mechanism of Action
Glofitamab-gxbm is a bispecific antibody that binds to CD20 expressed on the surface of B cells, and to CD3 receptor expressed on the surface of T cells. Glofitamab-gxbm causes T-cell activation and proliferation, secretion of cytokines, and the lysis of CD20-expressing B cells. Glofitamab-gxbm showed anti-tumor activity in vivo in mouse models of DLBCL.
5.5 Embryo Fetal Toxicity
Based on its mechanism of action, COLUMVI may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with COLUMVI and for 1 month after the last dose [see Use in Specific Populations (8.1, 8.3)].
5 Warnings and Precautions
- Neurologic Toxicity: Can cause serious neurologic toxicity, including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS). Monitor for neurologic toxicity; withhold or permanently discontinue based on severity. (5.2)
- Serious Infections: Can cause serious or fatal infections. Monitor patients for signs and symptoms of infection and treat appropriately. (5.3)
- Tumor Flare: Can cause serious tumor flare reactions. Monitor patients at risk for complications of tumor flare. (5.4)
- Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception. (5.5, 8.1, 8.3)
2 Dosage and Administration
- Pretreat with a single 1,000 mg dose of obinutuzumab intravenously 7 days before initiation of COLUMVI (Cycle 1 Day 1). (2.2)
- Administer premedications as recommended. (2.3)
- Administer only as an intravenous infusion. (2.1)
- Recommended dosage (2.2):
Treatment Cycle Cycle = 21 daysDay Dose of COLUMVI Day 1 Obinutuzumab 1,000 mg Cycle 1 Day 8 Step-up dose 1 2.5 mg Day 15 Step-up dose 2 10 mg Cycle 2 to 12 Day 1 30 mg - Administer in a facility equipped to monitor and manage CRS. (2.1, 2.2)
- Patients should be hospitalized for the 2.5 mg step-up dose and for subsequent infusions as recommended. (2.1, 2.2)
- See Full Prescribing Information for instructions on preparation and administration. (2.5, 2.6, 2.7)
3 Dosage Forms and Strengths
Injection:
- 2.5 mg/2.5 mL (1 mg/mL) clear, colorless solution in a single-dose vial.
- 10 mg/10 mL (1 mg/mL) clear, colorless solution in a single-dose vial.
5.1 Cytokine Release Syndrome
COLUMVI can cause serious and fatal cytokine release syndrome (CRS) [see Adverse Reactions (6.1)].
Among 145 patients who received COLUMVI, CRS occurred in 70%, with Grade 1 CRS developing in 52% of all patients, Grade 2 in 14%, Grade 3 in 2.8%, and Grade 4 in 1.4%. The most common manifestations of CRS included fever, tachycardia, hypotension, chills, and hypoxia.
CRS occurred in 56% of patients after the 2.5 mg dose of COLUMVI, 35% after the 10 mg dose, 29% after the initial 30 mg target dose, and 2.8% after subsequent doses. With the first step-up dose of COLUMVI, the median time to onset of CRS (from the start of infusion) was 14 hours (range: 5 to 74 hours). CRS after any dose resolved in 98% of cases, with a median duration of CRS of 2 days (range: 1 to 14 days). Recurrent CRS occurred in 34% of all patients. CRS can first occur with the 10 mg dose; of 135 patients treated with the 10 mg dose of COLUMVI, 15 patients (11%) experienced their first CRS event with the 10 mg dose, of which 13 events were Grade 1, 1 event was Grade 2, and 1 event was Grade 3.
Administer COLUMVI in a facility equipped to monitor and manage CRS. Initiate therapy according to the COLUMVI step-up dosing schedule to reduce the risk of CRS, administer pretreatment medications, and ensure adequate hydration [Dosage and Administration (2.3)]. Patients should be hospitalized during and for 24 hours after completing infusion of the 2.5 mg step-up dose. Patients who experienced any grade CRS during the 2.5 mg step-up dose should be hospitalized during and for 24 hours after completion of the 10 mg step-up dose. For subsequent doses, patients who experienced Grade ≥ 2 CRS with the previous infusion should be hospitalized during and for 24 hours after the next COLUMVI infusion [see Dosage and Administration (2.1 and 2.2)].
At the first sign of CRS, immediately evaluate patients for hospitalization, manage per current practice guidelines, and administer supportive care; withhold or permanently discontinue COLUMVI based on severity [see Dosage and Administration (2.4)].
8 Use in Specific Populations
Lactation: Advise not to breastfeed. (8.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
2.1 Important Dosing Information
- Administer only as an intravenous infusion through a dedicated infusion line that includes a sterile 0.2-micron in-line filter.
- Administer COLUMVI diluted solution via intravenous bag infusion. The 2.5 mg dose may alternatively be administered via intravenous syringe infusion [see Dosage and Administration (2.5, 2.6, 2.7)].
- COLUMVI should only be administered by a healthcare professional with immediate access to appropriate medical support, including supportive medications to manage severe CRS [see Dosage and Administration (2.4)].
- Ensure adequate hydration before administering COLUMVI.
- Premedicate before each dose [see Dosage and Administration (2.3)].
- Following pretreatment with obinutuzumab, administer COLUMVI according to the step-up dosing schedule in Table 1 with appropriate premedication, including dexamethasone, to reduce the incidence and severity of CRS [see Dosage and Administration (2.3)].
- Due to the risk of CRS, patients should be hospitalized during and for 24 hours after completion of infusion of step-up dose 1 (2.5 mg on Cycle 1 Day 8) [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
- Patients who experienced any grade CRS during step-up dose 1 should be hospitalized during and for 24 hours after completion of step-up dose 2 (10 mg on Cycle 1 Day 15). CRS with step-up dose 2 can occur in patients who did not experience CRS with step-up dose 1 [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
- For subsequent doses, patients who experienced Grade ≥ 2 CRS with their previous infusion should be hospitalized during and for 24 hours after the completion of the next COLUMVI infusion.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Warning: Cytokine Release Syndrome
Cytokine Release Syndrome (CRS), including serious or fatal reactions, can occur in patients receiving COLUMVI. Premedicate before each dose, and initiate treatment with the COLUMVI step-up dosing schedule to reduce the risk of CRS. Withhold COLUMVI until CRS resolves or permanently discontinue based on severity [see Dosage and Administration (2.1, 2.2, 2.3, and 2.4) and Warnings and Precautions (5.1)] .
16 How Supplied/storage and Handling
COLUMVI (glofitamab-gxbm) injection is a sterile, preservative-free, colorless, clear solution for intravenous infusion.
COLUMVI is supplied as:
| Carton Contents | NDC |
|---|---|
| One 2.5 mg/2.5 mL (1 mg/mL) single-dose vial | NDC 50242-125-01 |
| One 10 mg/10 mL (1 mg/mL) single-dose vial | NDC 50242-127-01 |
2.5 Preparation Into An Intravenous Bag
This section describes preparation of all doses of COLUMVI into an intravenous bag. For preparation instructions for the 2.5 mg dose into an intravenous syringe, see subsection 2.6 [see Dosage and Administration (2.6)].
2.4 Dosage Modifications for Adverse Reactions
No dosage reduction for COLUMVI is recommended.
8.3 Females and Males of Reproductive Potential
COLUMVI may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
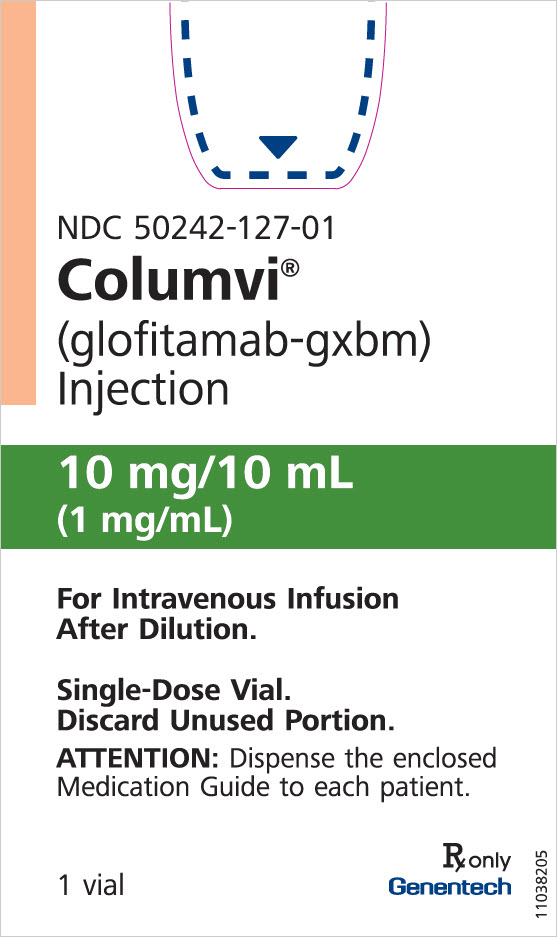
Principal Display Panel 10 Mg/10 Ml Vial Carton
NDC 50242-127-01
Columvi®
(glofitamab-gxbm)
Injection
10 mg/10 mL
(1 mg/mL)
For Intravenous Infusion
After Dilution.
Single-Dose Vial.
Discard Unused Portion.
ATTENTION: Dispense the enclosed
Medication Guide to each patient.
1 vial
Rx only
Genentech
11038205
Principal Display Panel 2.5 Mg/2.5 Ml Vial Carton
NDC 50242-125-01
Columvi®
(glofitamab-gxbm)
Injection
2.5 mg/2.5 mL
(1 mg/mL)
For Intravenous Infusion
After Dilution.
Single-Dose Vial.
Discard Unused Portion.
ATTENTION: Dispense the enclosed
Medication Guide to each patient.
1 vial
Rx only
Genentech
11038250
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or genotoxicity studies have been conducted with glofitamab-gxbm.
Fertility studies have not been conducted with glofitamab-gxbm.
2.6 Preparation of 2.5 Mg Dose Into An Intravenous Syringe
This section describes the alternative method of preparation of the 2.5 mg dose of COLUMVI into an intravenous syringe. For preparation instructions for all doses into an intravenous infusion bag, see subsection 2.5 [see Dosage and Administration (2.5)].
14.1 Relapsed Or Refractory Dlbcl, Nos Or Lbcl Arising From Follicular Lymphoma
The efficacy of COLUMVI was evaluated in Study NP30179 (NCT03075696), an open-label, multicenter, multicohort, single-arm clinical trial that included patients with relapsed or refractory LBCL after two or more lines of systemic therapy. The trial required an ECOG performance status of 0 or 1, absolute neutrophil count ≥ 1,500/µL, platelet count ≥ 75,000/µL independent of transfusion, serum creatinine ≤ 1.5 × ULN or CLcr ≥ 50 mL/min, and hepatic transaminases ≤ 3 × ULN. The trial excluded patients with active or previous CNS lymphoma or CNS disease, acute infection, recent infection requiring intravenous antibiotics, or prior allogeneic HSCT.
Following pretreatment with obinutuzumab on Cycle 1 Day 1, patients received COLUMVI by intravenous infusion, starting with a 2.5 mg step-up dose on Cycle 1 Day 8, followed by a 10 mg step-up dose on Cycle 1 Day 15, then 30 mg on Cycle 2 Day 1 and on Day 1 of each subsequent cycle. The cycle length was 21 days. COLUMVI was administered for up to 12 cycles unless patients experienced progressive disease or unacceptable toxicity.
The efficacy population consists of 132 patients with de novo DLBCL, NOS (80%) or LBCL arising from follicular lymphoma (20%) who received at least one dose of COLUMVI. The median age was 67 years (range: 21 to 90 years), 64% were male, 77% were White, 4.5% were Asian, 0.8% were Black or African American, 5% were Hispanic or Latino. The median number of prior lines of systemic therapy was 3 (range: 2 to 7). Most patients (83%) had refractory disease to the last therapy, 55% had primary refractory disease, 30% had received CAR-T cell therapy, and 19% had received autologous HSCT.
Efficacy was based on objective response rate (ORR) and duration of response (DOR), as determined by an Independent Review Committee (IRC) using the 2014 Lugano criteria.
Efficacy results are summarized in Table 12. The median time to first response was 42 days (range: 31 to 178 days). Among responders, the estimated median follow-up for DOR was 11.6 months.
| Outcome per IRC | COLUMVI N=132 |
|---|---|
| CI = confidence interval; NE = not estimable | |
| Overall Response Rate, n (%) | 74 (56) |
| (95% CI) | (47, 65) |
| Complete Response, n (%) | 57 (43) |
| (95% CI) | (35, 52) |
| Partial Response, n (%) | 17 (13) |
| (95% CI) | (8, 20) |
|
Duration of Response
From date of first response (PR or CR) until disease progression or death due to any cause.
|
N = 74 |
| Median DOR, months (95% CI) Kaplan-Meier estimate.
|
18.4 (11.4, NE) |
| 9-month estimate, % (95% CI) | 68.5 (56.7, 80.3) |
Structured Label Content
Section 42229-5 (42229-5)
Pretreatment with Obinutuzumab
Pretreat all patients with a single 1,000 mg dose of obinutuzumab administered as an intravenous infusion on Cycle 1 Day 1, 7 days prior to initiation of COLUMVI (see Table 1) to deplete the circulating and lymphoid tissue B cells.
Obinutuzumab should be administered as an intravenous infusion at 50 mg/hour. The rate of infusion can be escalated in 50 mg/hour increments every 30 minutes to a maximum of 400 mg/hour. Refer to the obinutuzumab prescribing information for complete dosing information.
Section 42231-1 (42231-1)
| MEDICATION GUIDE COLUMVI® (ko-loom-vee) (glofitamab-gxbm) injection, for intravenous infusion |
||
|---|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 6/2025 | |
|
What is the most important information I should know about COLUMVI? COLUMVI can cause Cytokine Release Syndrome (CRS), a serious side effect that is common during treatment with COLUMVI, and can also be serious and lead to death.
|
||
|
|
|
|
||
|
What is COLUMVI?
COLUMVI is a prescription medicine used to treat adults with certain types of diffuse large B-cell lymphoma (DLBCL) or large B-cell lymphoma (LBCL) that has come back (relapsed) or that did not respond to previous treatment (refractory), and who have received 2 or more prior treatments for their cancer. It is not known if COLUMVI is safe and effective in children. |
||
Before receiving COLUMVI, tell your healthcare provider about all of your medical conditions, including if you:
|
||
|
How will I receive COLUMVI?
|
||
|
What should I avoid while receiving COLUMVI? Do not drive, operate heavy machinery, or do other dangerous activities if you develop dizziness, confusion, shaking (tremors), sleepiness, or any other symptoms that impair consciousness until your signs and symptoms go away. These may be signs and symptoms of neurologic problems. See " What are the possible side effects of COLUMVI?" for more information about signs and symptoms of neurologic problems. |
||
|
What are the possible side effects of COLUMVI? COLUMVI may cause serious side effects, including:
|
||
|
|
|
|
||
| Your healthcare provider may temporarily stop or completely stop treatment with COLUMVI if you develop certain side effects. The most common side effects of COLUMVI include: CRS, muscle and bone pain, rash, and tiredness. The most common severe abnormal lab test results with COLUMVI include: decreased white blood cells, decreased phosphate (an electrolyte), increased uric acid levels, and decreased fibrinogen (a protein that helps with blood clotting). These are not all the possible side effects of COLUMVI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
|
General information about the safe and effective use of COLUMVI.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your healthcare provider or pharmacist for information about COLUMVI that is written for health professionals. |
||
|
What are the ingredients in COLUMVI? Active ingredient: glofitamab-gxbm Inactive ingredients: histidine, histidine hydrochloride monohydrate, methionine, polysorbate 20, sucrose, and Water for injection. Manufactured by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 U.S. License No.: 1048 For more information, go to www.COLUMVI.com or call 1-877-436-3683. |
Section 44425-7 (44425-7)
Store refrigerated at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not freeze. Do not shake.
11 Description (11 DESCRIPTION)
Glofitamab-gxbm is a bispecific CD20-directed CD3 T-cell engager. It is a recombinant humanized anti-CD20 anti-CD3ɛ bispecific immunoglobulin G1 (IgG1) monoclonal antibody produced in Chinese hamster ovary (CHO) cells. Glofitamab-gxbm has an approximate molecular weight of 197 kDa.
COLUMVI (glofitamab-gxbm) injection is a sterile, preservative-free, colorless, clear solution supplied in single-dose vials for intravenous infusion.
COLUMVI is supplied in 2.5 mg/2.5 mL and 10 mg/10 mL single-dose vials at a concentration of 1 mg/mL. Each mL of solution contains 1 mg glofitamab-gxbm, histidine (0.63 mg), histidine hydrochloride monohydrate (3.34 mg), methionine (1.49 mg), polysorbate 20 (0.5 mg), sucrose (82.15 mg), and Water for Injection, USP, at pH 5.5.
5.4 Tumor Flare
COLUMVI can cause serious tumor flare [see Adverse Reactions (6.1)]. Manifestations include localized pain and swelling at the sites of the lymphoma lesions and/or dyspnea from new pleural effusions.
Tumor flare was reported in 12% of patients who received COLUMVI, including Grade 2 tumor flare in 4.8% of patients and Grade 3 tumor flare in 2.8%. Recurrent tumor flare occurred in two (12%) of the affected patients. Most tumor flare events occurred during Cycle 1, with a median time to first onset of 2 days (range: 1 to 16 days) after the first dose of COLUMVI. The median duration was 3.5 days (range: 1 to 35 days).
Patients with bulky tumors or disease located in close proximity to airways or a vital organ should be monitored closely during initial therapy. Monitor for signs and symptoms of compression or obstruction due to mass effect secondary to tumor flare, and institute appropriate treatment. Withhold COLUMVI until tumor flare resolves [see Dosage and Administration (2.4)].
8.4 Pediatric Use
The safety and efficacy of COLUMVI in pediatric patients have not been established.
8.5 Geriatric Use
Of the 145 patients with relapsed or refractory LBCL who received COLUMVI in study NP30179, 55% were 65 years of age or older, and 23% were 75 years of age or older. There was a higher rate of fatal adverse reactions, primarily from COVID-19, in patients 65 years of age or older compared to younger patients [see Adverse Reactions (6.1)]. No overall differences in efficacy were observed between patients 65 years of age or older and younger patients.
12.6 Immunogenicity
The observed incidence of antidrug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the study described below with the incidence of ADA in other studies, including those of glofitamab-gxbm.
During treatment in Study NP30179 (up to 9 months) [see Clinical Studies (14.1)], using an enzyme-linked immunosorbent assay (ELISA), the incidence of anti-glofitamab antibody formation was 1.1% (5/448) in patients treated with COLUMVI. Because of the low occurrence of ADAs, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of glofitamab-gxbm is unknown.
4 Contraindications (4 CONTRAINDICATIONS)
None.
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Cytokine Release Syndrome [see Warnings and Precautions (5.1)]
- Neurologic Toxicity [see Warnings and Precautions (5.2)]
- Serious Infections [see Warnings and Precautions (5.3)]
- Tumor Flare [see Warnings and Precautions (5.4)]
7 Drug Interactions (7 DRUG INTERACTIONS)
For certain CYP substrates where minimal concentration changes may lead to serious adverse reactions, monitor for toxicities or drug concentrations of such CYP substrates when coadministered with COLUMVI.
Glofitamab-gxbm causes the release of cytokines [see Clinical Pharmacology (12.2)] that may suppress the activity of CYP enzymes, resulting in increased exposure of CYP substrates. Increased exposure of CYP substrates is more likely to occur after the first dose of COLUMVI on Cycle 1 Day 8 and up to 14 days after the first 30 mg dose on Cycle 2 Day 1 and during and after CRS [see Warnings and Precautions (5.1)].
12.3 Pharmacokinetics
The pharmacokinetics of glofitamab-gxbm was determined following pretreatment with a single dose of obinutuzumab of 1,000 mg and the pharmacokinetic parameters are presented as geometric mean (CV%) unless otherwise specified. Glofitamab-gxbm exposure increased dose-proportionally over the dose range from 0.005 to 30 mg (0.000167 to 1 time the recommended treatment dosage). Glofitamab-gxbm exposure parameters are summarized in Table 11 for the approved recommended dosage of COLUMVI.
| AUCtau (day∙mcg/mL) | Cmax (mcg/mL) | Ctrough (mcg/mL) | |
|---|---|---|---|
| Data presented as geometric mean (CV%). AUCtau = area under the concentration-time curve over one 21-day cycle; Cmax = maximum glofitamab-gxbm concentration; Ctrough = glofitamab-gxbm concentration prior to next dose; CV = geometric coefficient of variation. | |||
| First full 30 mg dose | 44.5 (55%) | 9.41 (27%) | 0.52 (83%) |
| Steady state Steady state values are approximated at Cycle 6 (week 18). 30 mg dose |
48.6 (33%) | 9.44 (26%) | 0.59 (67%) |
5.3 Serious Infections
COLUMVI can cause serious or fatal infections [see Adverse Reactions (6.1)].
Serious infections were reported in 16% of patients, including Grade 3 or 4 infections in 10%, and fatal infections in 4.8% of patients. Grade 3 or higher infections reported in ≥ 2% of patients were COVID-19 infection (6%), including COVID-19 pneumonia, and sepsis (4.1%). Febrile neutropenia occurred in 3.4% of patients.
COLUMVI should not be administered to patients with an active infection. Administer antimicrobial prophylaxis according to guidelines. Monitor patients before and during COLUMVI treatment for infection and treat appropriately. Withhold or consider permanent discontinuation of COLUMVI based on severity [see Dosage and Administration (2.4)].
1 Indications and Usage (1 INDICATIONS AND USAGE)
COLUMVI is indicated for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma, not otherwise specified (DLBCL, NOS) or large B-cell lymphoma (LBCL) arising from follicular lymphoma, after two or more lines of systemic therapy.
This indication is approved under accelerated approval based on response rate and durability of response [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
5.2 Neurologic Toxicity
COLUMVI can cause serious and fatal neurologic toxicity, including Immune Effector Cell-Associated Neurotoxicity (ICANS) [see Adverse Reactions (6.1)].
Among 145 patients who received COLUMVI, the most frequent neurologic toxicities of any grade were headache (10%), peripheral neuropathy (8%), dizziness or vertigo (7%), and mental status changes (4.8%, including confusional state, cognitive disorder, disorientation, somnolence, and delirium). Grade 3 or higher neurologic adverse reactions occurred in 2.1% of patients and included somnolence, delirium, and myelitis. Cases of ICANS of any grade occurred in 4.8% of patients.
Coadministration of COLUMVI with other products that cause dizziness or mental status changes may increase the risk of neurologic toxicity. Optimize concomitant medications and hydration to avoid dizziness or mental status changes. Institute fall precautions as appropriate.
Monitor patients for signs and symptoms of neurologic toxicity, evaluate, and provide supportive therapy; withhold or permanently discontinue COLUMVI based on severity [see Dosage and Administration (2.4)].
Evaluate patients who experience neurologic toxicity such as tremors, dizziness, or adverse reactions that may impair cognition or consciousness promptly, including potential neurology evaluation. Advise affected patients to refrain from driving and/or engaging in hazardous occupations or activities, such as operating heavy or potentially dangerous machinery, until the neurologic toxicity fully resolves.
12.1 Mechanism of Action
Glofitamab-gxbm is a bispecific antibody that binds to CD20 expressed on the surface of B cells, and to CD3 receptor expressed on the surface of T cells. Glofitamab-gxbm causes T-cell activation and proliferation, secretion of cytokines, and the lysis of CD20-expressing B cells. Glofitamab-gxbm showed anti-tumor activity in vivo in mouse models of DLBCL.
5.5 Embryo Fetal Toxicity (5.5 Embryo-Fetal Toxicity)
Based on its mechanism of action, COLUMVI may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with COLUMVI and for 1 month after the last dose [see Use in Specific Populations (8.1, 8.3)].
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Neurologic Toxicity: Can cause serious neurologic toxicity, including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS). Monitor for neurologic toxicity; withhold or permanently discontinue based on severity. (5.2)
- Serious Infections: Can cause serious or fatal infections. Monitor patients for signs and symptoms of infection and treat appropriately. (5.3)
- Tumor Flare: Can cause serious tumor flare reactions. Monitor patients at risk for complications of tumor flare. (5.4)
- Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception. (5.5, 8.1, 8.3)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
- Pretreat with a single 1,000 mg dose of obinutuzumab intravenously 7 days before initiation of COLUMVI (Cycle 1 Day 1). (2.2)
- Administer premedications as recommended. (2.3)
- Administer only as an intravenous infusion. (2.1)
- Recommended dosage (2.2):
Treatment Cycle Cycle = 21 daysDay Dose of COLUMVI Day 1 Obinutuzumab 1,000 mg Cycle 1 Day 8 Step-up dose 1 2.5 mg Day 15 Step-up dose 2 10 mg Cycle 2 to 12 Day 1 30 mg - Administer in a facility equipped to monitor and manage CRS. (2.1, 2.2)
- Patients should be hospitalized for the 2.5 mg step-up dose and for subsequent infusions as recommended. (2.1, 2.2)
- See Full Prescribing Information for instructions on preparation and administration. (2.5, 2.6, 2.7)
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Injection:
- 2.5 mg/2.5 mL (1 mg/mL) clear, colorless solution in a single-dose vial.
- 10 mg/10 mL (1 mg/mL) clear, colorless solution in a single-dose vial.
5.1 Cytokine Release Syndrome
COLUMVI can cause serious and fatal cytokine release syndrome (CRS) [see Adverse Reactions (6.1)].
Among 145 patients who received COLUMVI, CRS occurred in 70%, with Grade 1 CRS developing in 52% of all patients, Grade 2 in 14%, Grade 3 in 2.8%, and Grade 4 in 1.4%. The most common manifestations of CRS included fever, tachycardia, hypotension, chills, and hypoxia.
CRS occurred in 56% of patients after the 2.5 mg dose of COLUMVI, 35% after the 10 mg dose, 29% after the initial 30 mg target dose, and 2.8% after subsequent doses. With the first step-up dose of COLUMVI, the median time to onset of CRS (from the start of infusion) was 14 hours (range: 5 to 74 hours). CRS after any dose resolved in 98% of cases, with a median duration of CRS of 2 days (range: 1 to 14 days). Recurrent CRS occurred in 34% of all patients. CRS can first occur with the 10 mg dose; of 135 patients treated with the 10 mg dose of COLUMVI, 15 patients (11%) experienced their first CRS event with the 10 mg dose, of which 13 events were Grade 1, 1 event was Grade 2, and 1 event was Grade 3.
Administer COLUMVI in a facility equipped to monitor and manage CRS. Initiate therapy according to the COLUMVI step-up dosing schedule to reduce the risk of CRS, administer pretreatment medications, and ensure adequate hydration [Dosage and Administration (2.3)]. Patients should be hospitalized during and for 24 hours after completing infusion of the 2.5 mg step-up dose. Patients who experienced any grade CRS during the 2.5 mg step-up dose should be hospitalized during and for 24 hours after completion of the 10 mg step-up dose. For subsequent doses, patients who experienced Grade ≥ 2 CRS with the previous infusion should be hospitalized during and for 24 hours after the next COLUMVI infusion [see Dosage and Administration (2.1 and 2.2)].
At the first sign of CRS, immediately evaluate patients for hospitalization, manage per current practice guidelines, and administer supportive care; withhold or permanently discontinue COLUMVI based on severity [see Dosage and Administration (2.4)].
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
Lactation: Advise not to breastfeed. (8.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
2.1 Important Dosing Information
- Administer only as an intravenous infusion through a dedicated infusion line that includes a sterile 0.2-micron in-line filter.
- Administer COLUMVI diluted solution via intravenous bag infusion. The 2.5 mg dose may alternatively be administered via intravenous syringe infusion [see Dosage and Administration (2.5, 2.6, 2.7)].
- COLUMVI should only be administered by a healthcare professional with immediate access to appropriate medical support, including supportive medications to manage severe CRS [see Dosage and Administration (2.4)].
- Ensure adequate hydration before administering COLUMVI.
- Premedicate before each dose [see Dosage and Administration (2.3)].
- Following pretreatment with obinutuzumab, administer COLUMVI according to the step-up dosing schedule in Table 1 with appropriate premedication, including dexamethasone, to reduce the incidence and severity of CRS [see Dosage and Administration (2.3)].
- Due to the risk of CRS, patients should be hospitalized during and for 24 hours after completion of infusion of step-up dose 1 (2.5 mg on Cycle 1 Day 8) [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
- Patients who experienced any grade CRS during step-up dose 1 should be hospitalized during and for 24 hours after completion of step-up dose 2 (10 mg on Cycle 1 Day 15). CRS with step-up dose 2 can occur in patients who did not experience CRS with step-up dose 1 [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
- For subsequent doses, patients who experienced Grade ≥ 2 CRS with their previous infusion should be hospitalized during and for 24 hours after the completion of the next COLUMVI infusion.
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Warning: Cytokine Release Syndrome (WARNING: CYTOKINE RELEASE SYNDROME)
Cytokine Release Syndrome (CRS), including serious or fatal reactions, can occur in patients receiving COLUMVI. Premedicate before each dose, and initiate treatment with the COLUMVI step-up dosing schedule to reduce the risk of CRS. Withhold COLUMVI until CRS resolves or permanently discontinue based on severity [see Dosage and Administration (2.1, 2.2, 2.3, and 2.4) and Warnings and Precautions (5.1)] .
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
COLUMVI (glofitamab-gxbm) injection is a sterile, preservative-free, colorless, clear solution for intravenous infusion.
COLUMVI is supplied as:
| Carton Contents | NDC |
|---|---|
| One 2.5 mg/2.5 mL (1 mg/mL) single-dose vial | NDC 50242-125-01 |
| One 10 mg/10 mL (1 mg/mL) single-dose vial | NDC 50242-127-01 |
2.5 Preparation Into An Intravenous Bag (2.5 Preparation into an Intravenous Bag)
This section describes preparation of all doses of COLUMVI into an intravenous bag. For preparation instructions for the 2.5 mg dose into an intravenous syringe, see subsection 2.6 [see Dosage and Administration (2.6)].
2.4 Dosage Modifications for Adverse Reactions
No dosage reduction for COLUMVI is recommended.
8.3 Females and Males of Reproductive Potential
COLUMVI may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Principal Display Panel 10 Mg/10 Ml Vial Carton (PRINCIPAL DISPLAY PANEL - 10 mg/10 mL Vial Carton)
NDC 50242-127-01
Columvi®
(glofitamab-gxbm)
Injection
10 mg/10 mL
(1 mg/mL)
For Intravenous Infusion
After Dilution.
Single-Dose Vial.
Discard Unused Portion.
ATTENTION: Dispense the enclosed
Medication Guide to each patient.
1 vial
Rx only
Genentech
11038205
Principal Display Panel 2.5 Mg/2.5 Ml Vial Carton (PRINCIPAL DISPLAY PANEL - 2.5 mg/2.5 mL Vial Carton)
NDC 50242-125-01
Columvi®
(glofitamab-gxbm)
Injection
2.5 mg/2.5 mL
(1 mg/mL)
For Intravenous Infusion
After Dilution.
Single-Dose Vial.
Discard Unused Portion.
ATTENTION: Dispense the enclosed
Medication Guide to each patient.
1 vial
Rx only
Genentech
11038250
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or genotoxicity studies have been conducted with glofitamab-gxbm.
Fertility studies have not been conducted with glofitamab-gxbm.
2.6 Preparation of 2.5 Mg Dose Into An Intravenous Syringe (2.6 Preparation of 2.5 mg Dose into an Intravenous Syringe)
This section describes the alternative method of preparation of the 2.5 mg dose of COLUMVI into an intravenous syringe. For preparation instructions for all doses into an intravenous infusion bag, see subsection 2.5 [see Dosage and Administration (2.5)].
14.1 Relapsed Or Refractory Dlbcl, Nos Or Lbcl Arising From Follicular Lymphoma (14.1 Relapsed or Refractory DLBCL, NOS or LBCL Arising from Follicular Lymphoma)
The efficacy of COLUMVI was evaluated in Study NP30179 (NCT03075696), an open-label, multicenter, multicohort, single-arm clinical trial that included patients with relapsed or refractory LBCL after two or more lines of systemic therapy. The trial required an ECOG performance status of 0 or 1, absolute neutrophil count ≥ 1,500/µL, platelet count ≥ 75,000/µL independent of transfusion, serum creatinine ≤ 1.5 × ULN or CLcr ≥ 50 mL/min, and hepatic transaminases ≤ 3 × ULN. The trial excluded patients with active or previous CNS lymphoma or CNS disease, acute infection, recent infection requiring intravenous antibiotics, or prior allogeneic HSCT.
Following pretreatment with obinutuzumab on Cycle 1 Day 1, patients received COLUMVI by intravenous infusion, starting with a 2.5 mg step-up dose on Cycle 1 Day 8, followed by a 10 mg step-up dose on Cycle 1 Day 15, then 30 mg on Cycle 2 Day 1 and on Day 1 of each subsequent cycle. The cycle length was 21 days. COLUMVI was administered for up to 12 cycles unless patients experienced progressive disease or unacceptable toxicity.
The efficacy population consists of 132 patients with de novo DLBCL, NOS (80%) or LBCL arising from follicular lymphoma (20%) who received at least one dose of COLUMVI. The median age was 67 years (range: 21 to 90 years), 64% were male, 77% were White, 4.5% were Asian, 0.8% were Black or African American, 5% were Hispanic or Latino. The median number of prior lines of systemic therapy was 3 (range: 2 to 7). Most patients (83%) had refractory disease to the last therapy, 55% had primary refractory disease, 30% had received CAR-T cell therapy, and 19% had received autologous HSCT.
Efficacy was based on objective response rate (ORR) and duration of response (DOR), as determined by an Independent Review Committee (IRC) using the 2014 Lugano criteria.
Efficacy results are summarized in Table 12. The median time to first response was 42 days (range: 31 to 178 days). Among responders, the estimated median follow-up for DOR was 11.6 months.
| Outcome per IRC | COLUMVI N=132 |
|---|---|
| CI = confidence interval; NE = not estimable | |
| Overall Response Rate, n (%) | 74 (56) |
| (95% CI) | (47, 65) |
| Complete Response, n (%) | 57 (43) |
| (95% CI) | (35, 52) |
| Partial Response, n (%) | 17 (13) |
| (95% CI) | (8, 20) |
|
Duration of Response
From date of first response (PR or CR) until disease progression or death due to any cause.
|
N = 74 |
| Median DOR, months (95% CI) Kaplan-Meier estimate.
|
18.4 (11.4, NE) |
| 9-month estimate, % (95% CI) | 68.5 (56.7, 80.3) |
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:47:45.951897 · Updated: 2026-03-14T22:49:12.025925