Balversa
2a8aa5c0-6c92-4566-8c45-e8f4d1fc20ee
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
BALVERSA is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) with susceptible FGFR3 genetic alterations whose disease has progressed on or after at least one line of prior systemic therapy. Select patients for therapy based on an FDA-approved companion diagnostic for BALVERSA [see Dosage and Administration (2.1) and Clinical Studies (14.1) ] .
Dosage and Administration
Confirm the presence of FGFR3 genetic alterations in tumor specimens prior to initiation of treatment with BALVERSA. ( 2.1 ) Recommended initial dosage: 8 mg orally once daily with a dose increase to 9 mg daily if criteria are met. ( 2.2 ) Swallow whole with or without food. ( 2.2 )
Contraindications
None.
Warnings and Precautions
Ocular disorders: BALVERSA can cause central serous retinopathy/retinal pigment epithelial detachment (CSR/RPED). Perform monthly ophthalmological examinations during the first four months of treatment, every 3 months afterwards, and at any time for visual symptoms. Withhold BALVERSA when CSR/RPED occurs and permanently discontinue if it does not resolve within 4 weeks or if Grade 4 in severity. ( 2.3 , 5.1 ) Hyperphosphatemia: Increases in phosphate levels are a pharmacodynamic effect of BALVERSA. Monitor for hyperphosphatemia and manage with dose modifications when required. ( 2.3 , 5.2 ) Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to the fetus and to use effective contraception ( 5.3 , 8.1 , 8.3)
Adverse Reactions
The recommended dose modifications for adverse reactions are listed in Table 1. Table 1: BALVERSA Dose Reduction Schedule Dose 1 st dose reduction 2 nd dose reduction 3 rd dose reduction 4 th dose reduction 5 th dose reduction 9 mg ➞ (three 3 mg tablets) 8 mg (two 4 mg tablets) 6 mg (two 3 mg tablets) 5 mg (one 5 mg tablet) 4 mg (one 4 mg tablet) Stop 8 mg ➞ (two 4 mg tablets) 6 mg (two 3 mg tablets) 5 mg (one 5 mg tablet) 4 mg (one 4 mg tablet) Stop Table 2 summarizes recommendations for dose interruption, reduction, or discontinuation of BALVERSA in the management of specific adverse reactions. Table 2: Dose Modifications for Adverse Reactions Adverse Reaction BALVERSA Dose Modification Hyperphosphatemia In all patients, restrict phosphate intake to 600–800 mg daily. <6.99 mg/dL Continue BALVERSA at current dose. 7–8.99 mg/dL Continue BALVERSA at current dose. Start phosphate binder with food until phosphate level is <7 mg/dL. Reduce the dose if serum phosphate remains ≥7 mg/dL for a period of 2 months or if clinically necessary. 9–10 mg/dL Withhold BALVERSA with weekly reassessments until level returns to <7 mg/dL. Then restart BALVERSA at the same dose level. Start phosphate binder with food until serum phosphate level returns to <7 mg/dL. Reduce the dose if serum phosphate remains ≥9 mg/dL for a period of 1 month or if clinically necessary. >10 mg/dL Withhold BALVERSA with weekly reassessments until level returns to <7 mg/dL. Then may restart BALVERSA at the first reduced dose level. If hyperphosphatemia (≥10 mg/dL) for >2 weeks, discontinue BALVERSA permanently. Medical management of symptoms as clinically relevant. Serum phosphate with life-threatening consequences; urgent intervention indicated (e.g., dialysis) Discontinue BALVERSA permanently. Central Serous Retinopathy (CSR) Any Withhold BALVERSA and perform an ophthalmic evaluation within 2 weeks: If improving within 14 days, restart BALVERSA at the current dose. If not improving within 14 days, withhold BALVERSA until improving; once improving, may resume at the next lower dose level. Upon restarting BALVERSA, monitor for recurrence every 1 to 2 weeks for a month. If recurs or has not improved after 4 weeks of withholding BALVERSA, consider permanent discontinuation. Other Adverse Reactions Dose adjustment graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAEv5.0). Grade 3 Withhold BALVERSA until resolves to Grade 1 or baseline, then may resume dose level lower. Grade 4 Permanently discontinue.
Drug Interactions
Moderate CYP2C9 or strong CYP3A4 inhibitors: Consider alternative agents or monitor closely for adverse reactions. ( 7.1 ) Strong CYP3A4 inducers: Avoid concomitant use with BALVERSA. ( 7.1 ) Moderate CYP3A4 inducers: Administer BALVERSA at a dose of 9 mg. ( 7.1 ) Serum phosphate level-altering agents: Avoid concomitant use with agents that can alter serum phosphate levels before the initial dose modification period. ( 2.3 , 7.1 ) P-gp substrates: Separate BALVERSA administration by at least 6 hours before or after administration of P-gp substrates with narrow therapeutic indices. ( 7.2 )
How Supplied
BALVERSA ® (erdafitinib) tablets are available in the strengths and packages listed below: 3 mg tablets: Yellow, round biconvex, film-coated, debossed with "3" on one side and "EF" on the other side. Bottle of 56-tablets with child resistant closure (NDC 59676-030-56). Bottle of 84-tablets with child resistant closure (NDC 59676-030-84). 4 mg tablets: Orange, round biconvex, film-coated, debossed with "4" on one side and "EF" on the other side. Bottle of 28-tablets with child resistant closure (NDC 59676-040-28). Bottle of 56-tablets with child resistant closure (NDC 59676-040-56). 5 mg tablets: Brown, round biconvex, film-coated, debossed with "5" on one side and "EF" on the other side. Bottle of 28-tablets with child resistant closure (NDC 59676-050-28).
Storage and Handling
BALVERSA ® (erdafitinib) tablets are available in the strengths and packages listed below: 3 mg tablets: Yellow, round biconvex, film-coated, debossed with "3" on one side and "EF" on the other side. Bottle of 56-tablets with child resistant closure (NDC 59676-030-56). Bottle of 84-tablets with child resistant closure (NDC 59676-030-84). 4 mg tablets: Orange, round biconvex, film-coated, debossed with "4" on one side and "EF" on the other side. Bottle of 28-tablets with child resistant closure (NDC 59676-040-28). Bottle of 56-tablets with child resistant closure (NDC 59676-040-56). 5 mg tablets: Brown, round biconvex, film-coated, debossed with "5" on one side and "EF" on the other side. Bottle of 28-tablets with child resistant closure (NDC 59676-050-28).
Description
BALVERSA is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) with susceptible FGFR3 genetic alterations whose disease has progressed on or after at least one line of prior systemic therapy. Select patients for therapy based on an FDA-approved companion diagnostic for BALVERSA [see Dosage and Administration (2.1) and Clinical Studies (14.1) ] .
Medication Information
Warnings and Precautions
Ocular disorders: BALVERSA can cause central serous retinopathy/retinal pigment epithelial detachment (CSR/RPED). Perform monthly ophthalmological examinations during the first four months of treatment, every 3 months afterwards, and at any time for visual symptoms. Withhold BALVERSA when CSR/RPED occurs and permanently discontinue if it does not resolve within 4 weeks or if Grade 4 in severity. ( 2.3 , 5.1 ) Hyperphosphatemia: Increases in phosphate levels are a pharmacodynamic effect of BALVERSA. Monitor for hyperphosphatemia and manage with dose modifications when required. ( 2.3 , 5.2 ) Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to the fetus and to use effective contraception ( 5.3 , 8.1 , 8.3)
Indications and Usage
BALVERSA is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) with susceptible FGFR3 genetic alterations whose disease has progressed on or after at least one line of prior systemic therapy. Select patients for therapy based on an FDA-approved companion diagnostic for BALVERSA [see Dosage and Administration (2.1) and Clinical Studies (14.1) ] .
Dosage and Administration
Confirm the presence of FGFR3 genetic alterations in tumor specimens prior to initiation of treatment with BALVERSA. ( 2.1 ) Recommended initial dosage: 8 mg orally once daily with a dose increase to 9 mg daily if criteria are met. ( 2.2 ) Swallow whole with or without food. ( 2.2 )
Contraindications
None.
Adverse Reactions
The recommended dose modifications for adverse reactions are listed in Table 1. Table 1: BALVERSA Dose Reduction Schedule Dose 1 st dose reduction 2 nd dose reduction 3 rd dose reduction 4 th dose reduction 5 th dose reduction 9 mg ➞ (three 3 mg tablets) 8 mg (two 4 mg tablets) 6 mg (two 3 mg tablets) 5 mg (one 5 mg tablet) 4 mg (one 4 mg tablet) Stop 8 mg ➞ (two 4 mg tablets) 6 mg (two 3 mg tablets) 5 mg (one 5 mg tablet) 4 mg (one 4 mg tablet) Stop Table 2 summarizes recommendations for dose interruption, reduction, or discontinuation of BALVERSA in the management of specific adverse reactions. Table 2: Dose Modifications for Adverse Reactions Adverse Reaction BALVERSA Dose Modification Hyperphosphatemia In all patients, restrict phosphate intake to 600–800 mg daily. <6.99 mg/dL Continue BALVERSA at current dose. 7–8.99 mg/dL Continue BALVERSA at current dose. Start phosphate binder with food until phosphate level is <7 mg/dL. Reduce the dose if serum phosphate remains ≥7 mg/dL for a period of 2 months or if clinically necessary. 9–10 mg/dL Withhold BALVERSA with weekly reassessments until level returns to <7 mg/dL. Then restart BALVERSA at the same dose level. Start phosphate binder with food until serum phosphate level returns to <7 mg/dL. Reduce the dose if serum phosphate remains ≥9 mg/dL for a period of 1 month or if clinically necessary. >10 mg/dL Withhold BALVERSA with weekly reassessments until level returns to <7 mg/dL. Then may restart BALVERSA at the first reduced dose level. If hyperphosphatemia (≥10 mg/dL) for >2 weeks, discontinue BALVERSA permanently. Medical management of symptoms as clinically relevant. Serum phosphate with life-threatening consequences; urgent intervention indicated (e.g., dialysis) Discontinue BALVERSA permanently. Central Serous Retinopathy (CSR) Any Withhold BALVERSA and perform an ophthalmic evaluation within 2 weeks: If improving within 14 days, restart BALVERSA at the current dose. If not improving within 14 days, withhold BALVERSA until improving; once improving, may resume at the next lower dose level. Upon restarting BALVERSA, monitor for recurrence every 1 to 2 weeks for a month. If recurs or has not improved after 4 weeks of withholding BALVERSA, consider permanent discontinuation. Other Adverse Reactions Dose adjustment graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAEv5.0). Grade 3 Withhold BALVERSA until resolves to Grade 1 or baseline, then may resume dose level lower. Grade 4 Permanently discontinue.
Drug Interactions
Moderate CYP2C9 or strong CYP3A4 inhibitors: Consider alternative agents or monitor closely for adverse reactions. ( 7.1 ) Strong CYP3A4 inducers: Avoid concomitant use with BALVERSA. ( 7.1 ) Moderate CYP3A4 inducers: Administer BALVERSA at a dose of 9 mg. ( 7.1 ) Serum phosphate level-altering agents: Avoid concomitant use with agents that can alter serum phosphate levels before the initial dose modification period. ( 2.3 , 7.1 ) P-gp substrates: Separate BALVERSA administration by at least 6 hours before or after administration of P-gp substrates with narrow therapeutic indices. ( 7.2 )
Storage and Handling
BALVERSA ® (erdafitinib) tablets are available in the strengths and packages listed below: 3 mg tablets: Yellow, round biconvex, film-coated, debossed with "3" on one side and "EF" on the other side. Bottle of 56-tablets with child resistant closure (NDC 59676-030-56). Bottle of 84-tablets with child resistant closure (NDC 59676-030-84). 4 mg tablets: Orange, round biconvex, film-coated, debossed with "4" on one side and "EF" on the other side. Bottle of 28-tablets with child resistant closure (NDC 59676-040-28). Bottle of 56-tablets with child resistant closure (NDC 59676-040-56). 5 mg tablets: Brown, round biconvex, film-coated, debossed with "5" on one side and "EF" on the other side. Bottle of 28-tablets with child resistant closure (NDC 59676-050-28).
How Supplied
BALVERSA ® (erdafitinib) tablets are available in the strengths and packages listed below: 3 mg tablets: Yellow, round biconvex, film-coated, debossed with "3" on one side and "EF" on the other side. Bottle of 56-tablets with child resistant closure (NDC 59676-030-56). Bottle of 84-tablets with child resistant closure (NDC 59676-030-84). 4 mg tablets: Orange, round biconvex, film-coated, debossed with "4" on one side and "EF" on the other side. Bottle of 28-tablets with child resistant closure (NDC 59676-040-28). Bottle of 56-tablets with child resistant closure (NDC 59676-040-56). 5 mg tablets: Brown, round biconvex, film-coated, debossed with "5" on one side and "EF" on the other side. Bottle of 28-tablets with child resistant closure (NDC 59676-050-28).
Description
BALVERSA is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) with susceptible FGFR3 genetic alterations whose disease has progressed on or after at least one line of prior systemic therapy. Select patients for therapy based on an FDA-approved companion diagnostic for BALVERSA [see Dosage and Administration (2.1) and Clinical Studies (14.1) ] .
Section 42229-5
Limitations of Use
BALVERSA is not recommended for the treatment of patients who are eligible for and have not received prior PD-1 or PD-L1 inhibitor therapy [see Clinical Studies (14.1)] .
Section 42230-3
| PATIENT INFORMATION
BALVERSA ®(bal-VER-sah) (erdafitinib) tablets |
|
|---|---|
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 10/2024 |
|
What is BALVERSA?
BALVERSA is a prescription medicine used to treat adults with bladder cancer (urothelial cancer) that has spread or cannot be removed by surgery:
BALVERSA is not recommended for the treatment of people who are eligible for and have not received prior PD-1 or PD-L1 inhibitor therapy. It is not known if BALVERSA is safe and effective in children. |
|
Before taking BALVERSA tell your healthcare provider about all of your medical conditions, including if you:
|
|
How should I take BALVERSA?
|
|
|
What are the possible side effects of BALVERSA?
BALVERSA may cause serious side effects, including:
|
|
|
|
| Tell your healthcare provider right away if you develop any nail or skin problems including nails separating from the nail bed, nail pain, nail bleeding, breaking of the nails, color or texture changes in your nails, infected skin around the nail, an itchy skin rash, dry skin, or cracks in the skin.
BALVERSA may affect fertility in females who are able to become pregnant. Talk to your healthcare provider if this is a concern for you. These are not all of the possible side effects of BALVERSA. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store BALVERSA?
|
|
|
General information about the safe and effective use of BALVERSA.
Medicines are sometimes prescribed for purposes other than those listed in Patient Information leaflets. Do not use BALVERSA for a condition for which it was not prescribed. Do not give BALVERSA to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about BALVERSA that is written for healthcare professionals. |
|
|
What are the ingredients in BALVERSA?
Active ingredient:erdafitinib Inactive ingredients: Tablet Core: Croscarmellose sodium, Magnesium stearate (from vegetable source), Mannitol, Meglumine, and Microcrystalline Cellulose. Film Coating (Opadry amb II): Glycerol monocaprylocaprate Type I, Polyvinyl alcohol-partially hydrolyzed, Sodium lauryl sulfate, Talc, Titanium dioxide, Iron oxide yellow, Iron oxide red (for the orange and brown tablets only), Ferrosoferric oxide/iron oxide black (for the brown tablets only). Manufactured for:Janssen Products, LP, Horsham, PA 19044, USA For patent information: www.janssenpatents.com © 2019 Janssen Pharmaceutical Companies For more information call Janssen Products, LP at 1-800-526-7736 (1-800-JANSSEN) or go to www.BALVERSA.com. |
Section 44425-7
Store at 20 °C to 25 °C (68 °F to 77 °F); excursions permitted between 15 °C to 30 °C (59 °F to 86 °F) [see USP Controlled Room Temperature].
11 Description
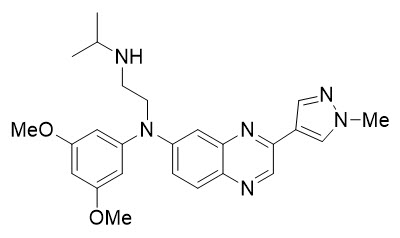
Erdafitinib, the active ingredient in BALVERSA, is a kinase inhibitor. The chemical name is N-(3,5-dimethoxyphenyl)-N'-(1-methylethyl)-N-[3-(1-methyl-1H-pyrazol-4-yl)quinoxalin-6-yl]ethane-1,2-diamine. Erdafitinib is a yellow powder. It is practically insoluble, or insoluble to freely soluble in organic solvents, and slightly soluble to practically insoluble, or insoluble in aqueous media over a wide range of pH values. The molecular formula is C 25H 30N 6O 2and molecular weight is 446.56.
Chemical structure of erdafitinib is as follows:
BALVERSA ®(erdafitinib) tablets are supplied as 3 mg, 4 mg or 5 mg film-coated tablets for oral administration and contains the following inactive ingredients:
Tablet Core: Croscarmellose sodium, Magnesium stearate (from vegetable source), Mannitol, Meglumine, and Microcrystalline Cellulose.
Film Coating: (Opadry amb II): Glycerol monocaprylocaprate Type I, Polyvinyl alcohol-partially hydrolyzed, Sodium lauryl sulfate, Talc, Titanium dioxide, Iron oxide yellow, Iron oxide red (for the orange and brown tablets only), Ferrosoferric oxide/iron oxide black (for the brown tablets only).
8.4 Pediatric Use
Safety and effectiveness of BALVERSA in pediatric patients have not been established.
Skeletal adverse reactions have occurred in pediatric patients treated with BALVERSA. In a study of BALVERSA that included pediatric patients ages 6 to <18 years with FGFR-positive advanced solid tumors, epiphysiolysis and bone fractures occurred.
In the postmarket setting and in literature reports, cases of slipped capital femoral epiphysis and accelerated linear growth in patients treated with BALVERSA have been reported.
8.5 Geriatric Use
Of the 479 patients treated with BALVERSA in clinical studies, 40% of patients were less than 65 years old, 40% of patients were 65 years to 74 years old, and 20% were 75 years old and over.
Patients 65 years of age and older treated with BALVERSA experienced a higher incidence of adverse reactions requiring treatment discontinuation than younger patients. In clinical trials, the incidence of treatment discontinuations of BALVERSA due to adverse reactions was 10% in patients younger than 65 years, 20% in patients ages 65–74 years, and 35% in patients 75 years or older.
No overall difference in efficacy was observed between these patients and younger patients [see Clinical Studies (14.1)].
4 Contraindications
None.
6 Adverse Reactions
The following serious adverse reactions are also described elsewhere in the labeling:
- Ocular Disorders [see Warnings and Precautions (5.1)] .
- Hyperphosphatemia [see Warnings and Precautions (5.2)] .
7 Drug Interactions
- Moderate CYP2C9 or strong CYP3A4 inhibitors: Consider alternative agents or monitor closely for adverse reactions. ( 7.1)
- Strong CYP3A4 inducers: Avoid concomitant use with BALVERSA. ( 7.1)
- Moderate CYP3A4 inducers: Administer BALVERSA at a dose of 9 mg. ( 7.1)
- Serum phosphate level-altering agents: Avoid concomitant use with agents that can alter serum phosphate levels before the initial dose modification period. ( 2.3, 7.1)
- P-gp substrates: Separate BALVERSA administration by at least 6 hours before or after administration of P-gp substrates with narrow therapeutic indices. ( 7.2)
5.1 Ocular Disorders
BALVERSA can cause ocular disorders, including central serous retinopathy/retinal pigment epithelial detachment (CSR/RPED) resulting in visual field defect.
In the pooled safety population [see Adverse Reactions (6)] , CSR/RPED occurred in 22% of patients treated with BALVERSA, with a median time to first onset of 46 days. In 104 patients with CSR, 40% required dose interruptions and 56% required dose reductions; 2.9% of BALVERSA-treated patients required permanent discontinuation for CSR. Of the 24 patients who restarted BALVERSA after dose interruption with or without dose reduction, 67% had recurrence and/or worsening of CSR after restarting. CSR was ongoing in 41% of the 104 patients at the time of last evaluation.
Dry eye symptoms occurred in 26% of BALVERSA-treated patients. All patients should receive dry eye prophylaxis with ocular demulcents as needed.
Perform monthly ophthalmological examinations during the first 4 months of treatment and every 3 months afterwards, and urgently at any time for visual symptoms. Ophthalmological examination should include assessment of visual acuity, slit lamp examination, fundoscopy, and optical coherence tomography.
Withhold or permanently discontinue BALVERSA based on severity and/or ophthalmology exam findings [see Dosage and Administration (2.3)] .
12.3 Pharmacokinetics
Following administration of BALVERSA 8 mg once daily, the mean (coefficient of variation [CV%]) erdafitinib steady-state maximum plasma concentration (C max), area under the curve (AUC tau), and minimum plasma concentration (C min) were 1,399 ng/mL (51%), 29,268 ng∙h/mL (60%), and 936 ng/mL (65%), respectively.
Following single and repeat once daily dosing of BALVERSA, erdafitinib exposure (C maxand AUC) increased proportionally across the dose range of 0.5 to 12 mg (0.06 to 1.3 times the maximum approved recommended dose). Steady state was achieved after 2 weeks with once daily dosing with a mean accumulation ratio was 4-fold.
12.5 Pharmacogenomics
CYP2C9 activity is reduced in individuals with genetic variants, such as the CYP2C9*2 and CYP2C9*3 polymorphisms. Erdafitinib exposure was similar in subjects with CYP2C9*1/*2 and *1/*3 genotypes relative to subjects with CYP2C9*1/*1 genotype (wild type). No data are available in subjects characterized by other genotypes (e.g., *2/*2, *2/*3, *3/*3). Simulation suggested no clinically meaningful differences in erdafitinib exposure in subjects with CYP2C9*2/*2 and *2/*3 genotypes. The exposure of erdafitinib is predicted to be 50% higher in subjects with the CYP2C9*3/*3 genotype, estimated to be present in 0.4% to 3% of the population among various ethnic groups.
2.1 Patient Selection
Select patients for the treatment of locally advanced or metastatic urothelial carcinoma with BALVERSA based on the presence of susceptible FGFR3genetic alterations in tumor specimens as detected by an FDA-approved companion diagnostic [see Clinical Studies (14.1)] .
Information on FDA-approved tests for the detection of FGFR3genetic alterations in urothelial cancer is available at: http://www.fda.gov/CompanionDiagnostics.
1 Indications and Usage
BALVERSA is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) with susceptible FGFR3 genetic alterations whose disease has progressed on or after at least one line of prior systemic therapy.
Select patients for therapy based on an FDA-approved companion diagnostic for BALVERSA [see Dosage and Administration (2.1) and Clinical Studies (14.1)] .
12.1 Mechanism of Action
Erdafitinib is a kinase inhibitor that binds to and inhibits enzymatic activity of FGFR1, FGFR2, FGFR3 and FGFR4 based on in vitro data. Erdafitinib inhibited FGFR phosphorylation and signaling and decreased cell viability in cell lines expressing FGFR genetic alterations, including point mutations, amplifications, and fusions. Erdafitinib demonstrated antitumor activity in FGFR-expressing cell lines and xenograft models derived from tumor types, including bladder cancer.
5.3 Embryo Fetal Toxicity
Based on the mechanism of action and findings in animal reproduction studies, BALVERSA can cause fetal harm when administered to a pregnant woman. In an embryo-fetal toxicity study, oral administration of erdafitinib to pregnant rats during the period of organogenesis caused malformations and embryo-fetal death at maternal exposures that were less than the human exposures at the maximum human recommended dose based on area under the curve (AUC). Advise pregnant women of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with BALVERSA and for one month after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with BALVERSA and for one month after the last dose [see Use in Specific Populations (8.1, 8.3)and Clinical Pharmacology (12.1)] .
5 Warnings and Precautions
- Ocular disorders: BALVERSA can cause central serous retinopathy/retinal pigment epithelial detachment (CSR/RPED). Perform monthly ophthalmological examinations during the first four months of treatment, every 3 months afterwards, and at any time for visual symptoms. Withhold BALVERSA when CSR/RPED occurs and permanently discontinue if it does not resolve within 4 weeks or if Grade 4 in severity. ( 2.3, 5.1)
- Hyperphosphatemia: Increases in phosphate levels are a pharmacodynamic effect of BALVERSA. Monitor for hyperphosphatemia and manage with dose modifications when required. ( 2.3, 5.2)
- Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to the fetus and to use effective contraception ( 5.3, 8.1, 8.3)
2 Dosage and Administration
3 Dosage Forms and Strengths
Tablets: 3 mg, 4 mg, and 5 mg. ( 3)
8 Use in Specific Populations
- Lactation: Advise not to breastfeed. ( 8.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflects exposure to BALVERSA as a single agent at the recommended dose (8 to 9 mg orally daily) in 479 patients with advanced urothelial cancer and FGFRalterations in 42756493BLC3001 (NCT03390504), 42756493BLC2001 (NCT02365597), 42756493BLC2002 (NCT 03473743), and 42756493EDI1001 (NCT01703481). Among 479 patients who received BALVERSA, the median duration of treatment was 4.8 months (range: 0.1 to 43 months). In this pooled safety population, the most common (>20%) adverse reactions, including laboratory abnormalities, were increased phosphate, nail disorders, stomatitis, diarrhea, increased creatinine, increased alkaline phosphatase, increased alanine aminotransferase, decreased hemoglobin, decreased sodium, increased aspartate aminotransferase, fatigue, dry mouth, dry skin, decreased phosphate, decreased appetite, dysgeusia, constipation, increased calcium, dry eye, palmar-plantar erythrodysesthesia syndrome, increased potassium, alopecia, and central serous retinopathy.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
2.2 Recommended Dosage and Schedule
The recommended starting dose of BALVERSA is 8 mg (two 4 mg tablets) orally once daily, with a dose increase to 9 mg (three 3 mg tablets) once daily based on tolerability, including hyperphosphatemia, at 14 to 21 days [see Dosage and Administration (2.3)].
Swallow tablets whole with or without food. If vomiting occurs any time after taking BALVERSA, the next dose should be taken the next day. Treatment should continue until disease progression or unacceptable toxicity occurs.
If a dose of BALVERSA is missed, it can be taken as soon as possible on the same day. Resume the regular daily dose schedule for BALVERSA the next day. Extra tablets should not be taken to make up for the missed dose.
16 How Supplied/storage and Handling
BALVERSA ®(erdafitinib) tablets are available in the strengths and packages listed below:
- 3 mg tablets: Yellow, round biconvex, film-coated, debossed with "3" on one side and "EF" on the other side.
- Bottle of 56-tablets with child resistant closure (NDC 59676-030-56).
- Bottle of 84-tablets with child resistant closure (NDC 59676-030-84).
- 4 mg tablets: Orange, round biconvex, film-coated, debossed with "4" on one side and "EF" on the other side.
- Bottle of 28-tablets with child resistant closure (NDC 59676-040-28).
- Bottle of 56-tablets with child resistant closure (NDC 59676-040-56).
- 5 mg tablets: Brown, round biconvex, film-coated, debossed with "5" on one side and "EF" on the other side.
- Bottle of 28-tablets with child resistant closure (NDC 59676-050-28).
7.1 Effect of Other Drugs On Balversa
Table 7 summarizes drug interactions that affect the exposure of BALVERSA or serum phosphate level and their clinical management.
| Moderate CYP2C9 or Strong CYP3A4 Inhibitors | |
| Clinical Impact |
|
| Clinical Management |
|
| Strong CYP3A4 Inducers | |
| Clinical Impact |
|
| Clinical Management |
|
| Moderate CYP3A4 Inducers | |
| Clinical Impact |
|
| Clinical Management |
|
| Serum Phosphate Level-Altering Agents | |
| Clinical Impact |
|
| Clinical Management |
|
7.2 Effect of Balversa On Other Drugs
Table 8 summarizes the effect of BALVERSA on other drugs and their clinical management.
| P-glycoprotein (P-gp) Substrates | |
|---|---|
| Clinical Impact |
|
| Clinical Management |
|
2.3 Dose Modifications for Adverse Reactions
The recommended dose modifications for adverse reactions are listed in Table 1.
| Dose | 1 stdose reduction | 2 nddose reduction | 3 rddose reduction | 4 thdose reduction | 5 thdose reduction |
|---|---|---|---|---|---|
|
9 mg ➞
(three 3 mg tablets) |
8 mg
(two 4 mg tablets) |
6 mg
(two 3 mg tablets) |
5 mg
(one 5 mg tablet) |
4 mg
(one 4 mg tablet) |
Stop |
|
8 mg ➞
(two 4 mg tablets) |
6 mg
(two 3 mg tablets) |
5 mg
(one 5 mg tablet) |
4 mg
(one 4 mg tablet) |
Stop |
Table 2 summarizes recommendations for dose interruption, reduction, or discontinuation of BALVERSA in the management of specific adverse reactions.
| Adverse Reaction | BALVERSA Dose Modification |
|---|---|
| Hyperphosphatemia | |
| In all patients, restrict phosphate intake to 600–800 mg daily. | |
| <6.99 mg/dL | Continue BALVERSA at current dose. |
| 7–8.99 mg/dL |
|
| 9–10 mg/dL |
|
| >10 mg/dL |
|
| Serum phosphate with life-threatening consequences; urgent intervention indicated (e.g., dialysis) |
|
| Central Serous Retinopathy (CSR) | |
| Any | Withhold BALVERSA and perform an ophthalmic evaluation within 2 weeks:
If recurs or has not improved after 4 weeks of withholding BALVERSA, consider permanent discontinuation. |
|
Other Adverse Reactions
Dose adjustment graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAEv5.0).
|
|
| Grade 3 | Withhold BALVERSA until resolves to Grade 1 or baseline, then may resume dose level lower. |
| Grade 4 | Permanently discontinue. |
8.3 Females and Males of Reproductive Potential
BALVERSA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)] .
14.1 Urothelial Carcinoma With Susceptible Fgfr3
The efficacy of BALVERSA was evaluated in Study BLC3001 (NCT03390504) Cohort 1, a randomized, open-label, multicenter study in which 266 patients with advanced urothelial cancer harboring selected FGFR3alterations were randomized 1:1 to receive BALVERSA (8 mg with titration up to 9 mg) versus chemotherapy (docetaxel 75 mg/m 2once every 3 weeks or vinflunine 320 mg/m 2once every 3 weeks) until unacceptable toxicity or progression. Randomization was stratified by region (North America vs. Europe vs. rest of world), Eastern Cooperative Oncology Group (ECOG) performance status (0 or 1 vs. 2) and visceral or bone metastases (yes vs. no). All patients needed to have had disease progression after 1 or 2 prior treatments, at least 1 of which included a PD-1 or PD-L1 inhibitor. FGFR3genetic alterations were identified from tumor tissue in a central laboratory by the QIAGEN therascreen ® FGFRRGQ RT-Polymerase Chain Reaction (PCR) kit in 75% of patients while the remainder (25%) were identified by local next generation sequencing (NGS) assays.
The major efficacy outcome measures were overall survival (OS), progression-free survival (PFS), and objective response rate (ORR) assessed by investigator using RECIST (Response Evaluation Criteria in Solid Tumors) Version 1.1.
The median age was 67 years (range: 32 to 86 years) and 71% were male; 54% were White, 29% Asian, 0.4% Black, 0.4% multiple races, 16% not reported; 2% were Hispanic/Latino; and baseline ECOG performance status was 0 (43%), 1 (48%), or 2 (9%). Eighty-one percent of patients had FGFR3mutations, 17% had fusions, and 2% had both mutations and fusions. Ninety-five percent of patients had pure transitional cell carcinoma (TCC) and 5% had TCC with other histologic variants. The primary tumor location was the upper tract for 33% of subjects and lower tract for 67%; 74% of patients had visceral or bone metastases. Eighty-eight percent of patients received platinum-containing chemotherapy previously. PD-1 or PD-L1 inhibitor therapy was received only in the neoadjuvant or adjuvant setting in 7% of patients.
Statistically significant improvements in OS, PFS, and ORR were demonstrated for BALVERSA compared with chemotherapy.
Table 9 and Figures 1 and 2 summarize the efficacy results for BLC3001 Cohort 1.
| BALVERSA
N=136 |
Chemotherapy
N=130 |
|
|---|---|---|
| All p-values reported are 2-sided and compared with 0.019 of the allocated alpha for the interim analysis.
ORR = confirmed objective response (CR + PR) CI = Confidence Interval |
||
| Overall Survival (OS) | ||
| Number of events (%) | 77 (56.6%) | 78 (60.0%) |
| Median
Based on Kaplan-Meier estimates , months (95% CI)
|
12.1 (10.3, 16.4) | 7.8 (6.5, 11.1) |
| Hazard ratio
Based on an unstratified Cox proportional hazard model (95% CI)
|
0.64 (0.47, 0.88) | |
| p-value
Based on an unstratified log-rank test
|
0.0050 | |
| Progression-free survival (PFS) | ||
| Number of events (%) | 101 (74.3%) | 90 (69.2%) |
| Median , months (95% CI) | 5.6 (4.4, 5.7) | 2.7 (1.8, 3.7) |
| Hazard ratio (95% CI) | 0.58 (0.44, 0.78) | |
| p-value | 0.0002 | |
| Objective response rate (ORR) | ||
| ORR (95% CI) | 35.3% (27.3, 43.9) | 8.5% (4.3, 14.6) |
| p-value
p-value is estimated using Cochran-Haenszel (CMH) test with ECOG performance status (0 or 1 vs 2) as a stratification factor.
|
<0.001 | |
| Complete response, CR (%) | 5.1% | 0.8% |
| Partial response, PR (%) | 30.1% | 7.7% |
Figure 1: Kaplan-Meier Plot of Overall Survival (Study BLC3001 Cohort 1)
Figure 2: Kaplan-Meier Plot of Progression-free Survival (Study BLC3001 Cohort 1)
Principal Display Panel 3 Mg Tablet Bottle Carton
NDC 59676-030-56
Balversa
®
(erdafitinib) tablets
3 mg
Each film-coated tablet
contains 3 mg of erdafitinib.
Rx only
56 film-coated tablets
Principal Display Panel 4 Mg Tablet Bottle Carton
NDC 59676-040-56
Balversa
®
(erdafitinib) tablets
4 mg
Each film-coated tablet
contains 4 mg of erdafitinib.
Rx only
56 film-coated tablets
Principal Display Panel 5 Mg Tablet Bottle Carton
NDC 59676-050-28
Balversa
®
(erdafitinib) tablets
5 mg
Each film-coated tablet
contains 5 mg of erdafitinib.
Rx only
28 film-coated tablets
5.2 Hyperphosphatemia and Soft Tissue Mineralization
BALVERSA can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcinosis, non-uremic calciphylaxis and vascular calcification. Increases in phosphate levels are a pharmacodynamic effect of BALVERSA [see Pharmacodynamics (12.2)].
In the pooled safety population [see Adverse Reactions (6)], increased phosphate occurred in 73% of BALVERSA-treated patients. The median onset time of increased phosphate was 16 days (range: 8–421) after initiating BALVERSA. Twenty-four percent of patients received phosphate binders during treatment with BALVERSA. Vascular calcification was observed in 0.2% of patients treated with BALVERSA.
Monitor for hyperphosphatemia throughout treatment. Restrict dietary phosphate intake (600–800 mg daily) and avoid concomitant use of agents that may increase serum phosphate levels.
If serum phosphate is above 7.0 mg/dL, consider adding an oral phosphate binder until serum phosphate level returns to <7.0 mg/dL. Withhold, dose reduce, or permanently discontinue BALVERSA based on duration and severity of hyperphosphatemia according to Table 2 [see Dosage and Administration (2.3)].
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenicity studies have not been conducted with erdafitinib.
Erdafitinib was not mutagenic in a bacterial reverse mutation (Ames) assay and was not clastogenic in an in vitro micronucleus or an in vivo rat bone marrow micronucleus assay.
Fertility studies in animals have not been conducted with erdafitinib. In the 3-month repeat-dose toxicity study, erdafitinib showed effects on female reproductive organs (necrosis of the ovarian corpora lutea) in rats at an exposure less than the human exposure (AUC) at maximum recommended human dose.
Structured Label Content
Section 42229-5 (42229-5)
Limitations of Use
BALVERSA is not recommended for the treatment of patients who are eligible for and have not received prior PD-1 or PD-L1 inhibitor therapy [see Clinical Studies (14.1)] .
Section 42230-3 (42230-3)
| PATIENT INFORMATION
BALVERSA ®(bal-VER-sah) (erdafitinib) tablets |
|
|---|---|
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 10/2024 |
|
What is BALVERSA?
BALVERSA is a prescription medicine used to treat adults with bladder cancer (urothelial cancer) that has spread or cannot be removed by surgery:
BALVERSA is not recommended for the treatment of people who are eligible for and have not received prior PD-1 or PD-L1 inhibitor therapy. It is not known if BALVERSA is safe and effective in children. |
|
Before taking BALVERSA tell your healthcare provider about all of your medical conditions, including if you:
|
|
How should I take BALVERSA?
|
|
|
What are the possible side effects of BALVERSA?
BALVERSA may cause serious side effects, including:
|
|
|
|
| Tell your healthcare provider right away if you develop any nail or skin problems including nails separating from the nail bed, nail pain, nail bleeding, breaking of the nails, color or texture changes in your nails, infected skin around the nail, an itchy skin rash, dry skin, or cracks in the skin.
BALVERSA may affect fertility in females who are able to become pregnant. Talk to your healthcare provider if this is a concern for you. These are not all of the possible side effects of BALVERSA. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store BALVERSA?
|
|
|
General information about the safe and effective use of BALVERSA.
Medicines are sometimes prescribed for purposes other than those listed in Patient Information leaflets. Do not use BALVERSA for a condition for which it was not prescribed. Do not give BALVERSA to other people, even if they have the same symptoms that you have. It may harm them. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about BALVERSA that is written for healthcare professionals. |
|
|
What are the ingredients in BALVERSA?
Active ingredient:erdafitinib Inactive ingredients: Tablet Core: Croscarmellose sodium, Magnesium stearate (from vegetable source), Mannitol, Meglumine, and Microcrystalline Cellulose. Film Coating (Opadry amb II): Glycerol monocaprylocaprate Type I, Polyvinyl alcohol-partially hydrolyzed, Sodium lauryl sulfate, Talc, Titanium dioxide, Iron oxide yellow, Iron oxide red (for the orange and brown tablets only), Ferrosoferric oxide/iron oxide black (for the brown tablets only). Manufactured for:Janssen Products, LP, Horsham, PA 19044, USA For patent information: www.janssenpatents.com © 2019 Janssen Pharmaceutical Companies For more information call Janssen Products, LP at 1-800-526-7736 (1-800-JANSSEN) or go to www.BALVERSA.com. |
Section 44425-7 (44425-7)
Store at 20 °C to 25 °C (68 °F to 77 °F); excursions permitted between 15 °C to 30 °C (59 °F to 86 °F) [see USP Controlled Room Temperature].
11 Description (11 DESCRIPTION)
Erdafitinib, the active ingredient in BALVERSA, is a kinase inhibitor. The chemical name is N-(3,5-dimethoxyphenyl)-N'-(1-methylethyl)-N-[3-(1-methyl-1H-pyrazol-4-yl)quinoxalin-6-yl]ethane-1,2-diamine. Erdafitinib is a yellow powder. It is practically insoluble, or insoluble to freely soluble in organic solvents, and slightly soluble to practically insoluble, or insoluble in aqueous media over a wide range of pH values. The molecular formula is C 25H 30N 6O 2and molecular weight is 446.56.
Chemical structure of erdafitinib is as follows:
BALVERSA ®(erdafitinib) tablets are supplied as 3 mg, 4 mg or 5 mg film-coated tablets for oral administration and contains the following inactive ingredients:
Tablet Core: Croscarmellose sodium, Magnesium stearate (from vegetable source), Mannitol, Meglumine, and Microcrystalline Cellulose.
Film Coating: (Opadry amb II): Glycerol monocaprylocaprate Type I, Polyvinyl alcohol-partially hydrolyzed, Sodium lauryl sulfate, Talc, Titanium dioxide, Iron oxide yellow, Iron oxide red (for the orange and brown tablets only), Ferrosoferric oxide/iron oxide black (for the brown tablets only).
8.4 Pediatric Use
Safety and effectiveness of BALVERSA in pediatric patients have not been established.
Skeletal adverse reactions have occurred in pediatric patients treated with BALVERSA. In a study of BALVERSA that included pediatric patients ages 6 to <18 years with FGFR-positive advanced solid tumors, epiphysiolysis and bone fractures occurred.
In the postmarket setting and in literature reports, cases of slipped capital femoral epiphysis and accelerated linear growth in patients treated with BALVERSA have been reported.
8.5 Geriatric Use
Of the 479 patients treated with BALVERSA in clinical studies, 40% of patients were less than 65 years old, 40% of patients were 65 years to 74 years old, and 20% were 75 years old and over.
Patients 65 years of age and older treated with BALVERSA experienced a higher incidence of adverse reactions requiring treatment discontinuation than younger patients. In clinical trials, the incidence of treatment discontinuations of BALVERSA due to adverse reactions was 10% in patients younger than 65 years, 20% in patients ages 65–74 years, and 35% in patients 75 years or older.
No overall difference in efficacy was observed between these patients and younger patients [see Clinical Studies (14.1)].
4 Contraindications (4 CONTRAINDICATIONS)
None.
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following serious adverse reactions are also described elsewhere in the labeling:
- Ocular Disorders [see Warnings and Precautions (5.1)] .
- Hyperphosphatemia [see Warnings and Precautions (5.2)] .
7 Drug Interactions (7 DRUG INTERACTIONS)
- Moderate CYP2C9 or strong CYP3A4 inhibitors: Consider alternative agents or monitor closely for adverse reactions. ( 7.1)
- Strong CYP3A4 inducers: Avoid concomitant use with BALVERSA. ( 7.1)
- Moderate CYP3A4 inducers: Administer BALVERSA at a dose of 9 mg. ( 7.1)
- Serum phosphate level-altering agents: Avoid concomitant use with agents that can alter serum phosphate levels before the initial dose modification period. ( 2.3, 7.1)
- P-gp substrates: Separate BALVERSA administration by at least 6 hours before or after administration of P-gp substrates with narrow therapeutic indices. ( 7.2)
5.1 Ocular Disorders
BALVERSA can cause ocular disorders, including central serous retinopathy/retinal pigment epithelial detachment (CSR/RPED) resulting in visual field defect.
In the pooled safety population [see Adverse Reactions (6)] , CSR/RPED occurred in 22% of patients treated with BALVERSA, with a median time to first onset of 46 days. In 104 patients with CSR, 40% required dose interruptions and 56% required dose reductions; 2.9% of BALVERSA-treated patients required permanent discontinuation for CSR. Of the 24 patients who restarted BALVERSA after dose interruption with or without dose reduction, 67% had recurrence and/or worsening of CSR after restarting. CSR was ongoing in 41% of the 104 patients at the time of last evaluation.
Dry eye symptoms occurred in 26% of BALVERSA-treated patients. All patients should receive dry eye prophylaxis with ocular demulcents as needed.
Perform monthly ophthalmological examinations during the first 4 months of treatment and every 3 months afterwards, and urgently at any time for visual symptoms. Ophthalmological examination should include assessment of visual acuity, slit lamp examination, fundoscopy, and optical coherence tomography.
Withhold or permanently discontinue BALVERSA based on severity and/or ophthalmology exam findings [see Dosage and Administration (2.3)] .
12.3 Pharmacokinetics
Following administration of BALVERSA 8 mg once daily, the mean (coefficient of variation [CV%]) erdafitinib steady-state maximum plasma concentration (C max), area under the curve (AUC tau), and minimum plasma concentration (C min) were 1,399 ng/mL (51%), 29,268 ng∙h/mL (60%), and 936 ng/mL (65%), respectively.
Following single and repeat once daily dosing of BALVERSA, erdafitinib exposure (C maxand AUC) increased proportionally across the dose range of 0.5 to 12 mg (0.06 to 1.3 times the maximum approved recommended dose). Steady state was achieved after 2 weeks with once daily dosing with a mean accumulation ratio was 4-fold.
12.5 Pharmacogenomics
CYP2C9 activity is reduced in individuals with genetic variants, such as the CYP2C9*2 and CYP2C9*3 polymorphisms. Erdafitinib exposure was similar in subjects with CYP2C9*1/*2 and *1/*3 genotypes relative to subjects with CYP2C9*1/*1 genotype (wild type). No data are available in subjects characterized by other genotypes (e.g., *2/*2, *2/*3, *3/*3). Simulation suggested no clinically meaningful differences in erdafitinib exposure in subjects with CYP2C9*2/*2 and *2/*3 genotypes. The exposure of erdafitinib is predicted to be 50% higher in subjects with the CYP2C9*3/*3 genotype, estimated to be present in 0.4% to 3% of the population among various ethnic groups.
2.1 Patient Selection
Select patients for the treatment of locally advanced or metastatic urothelial carcinoma with BALVERSA based on the presence of susceptible FGFR3genetic alterations in tumor specimens as detected by an FDA-approved companion diagnostic [see Clinical Studies (14.1)] .
Information on FDA-approved tests for the detection of FGFR3genetic alterations in urothelial cancer is available at: http://www.fda.gov/CompanionDiagnostics.
1 Indications and Usage (1 INDICATIONS AND USAGE)
BALVERSA is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma (mUC) with susceptible FGFR3 genetic alterations whose disease has progressed on or after at least one line of prior systemic therapy.
Select patients for therapy based on an FDA-approved companion diagnostic for BALVERSA [see Dosage and Administration (2.1) and Clinical Studies (14.1)] .
12.1 Mechanism of Action
Erdafitinib is a kinase inhibitor that binds to and inhibits enzymatic activity of FGFR1, FGFR2, FGFR3 and FGFR4 based on in vitro data. Erdafitinib inhibited FGFR phosphorylation and signaling and decreased cell viability in cell lines expressing FGFR genetic alterations, including point mutations, amplifications, and fusions. Erdafitinib demonstrated antitumor activity in FGFR-expressing cell lines and xenograft models derived from tumor types, including bladder cancer.
5.3 Embryo Fetal Toxicity (5.3 Embryo-Fetal Toxicity)
Based on the mechanism of action and findings in animal reproduction studies, BALVERSA can cause fetal harm when administered to a pregnant woman. In an embryo-fetal toxicity study, oral administration of erdafitinib to pregnant rats during the period of organogenesis caused malformations and embryo-fetal death at maternal exposures that were less than the human exposures at the maximum human recommended dose based on area under the curve (AUC). Advise pregnant women of the potential risk to the fetus. Advise female patients of reproductive potential to use effective contraception during treatment with BALVERSA and for one month after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with BALVERSA and for one month after the last dose [see Use in Specific Populations (8.1, 8.3)and Clinical Pharmacology (12.1)] .
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Ocular disorders: BALVERSA can cause central serous retinopathy/retinal pigment epithelial detachment (CSR/RPED). Perform monthly ophthalmological examinations during the first four months of treatment, every 3 months afterwards, and at any time for visual symptoms. Withhold BALVERSA when CSR/RPED occurs and permanently discontinue if it does not resolve within 4 weeks or if Grade 4 in severity. ( 2.3, 5.1)
- Hyperphosphatemia: Increases in phosphate levels are a pharmacodynamic effect of BALVERSA. Monitor for hyperphosphatemia and manage with dose modifications when required. ( 2.3, 5.2)
- Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to the fetus and to use effective contraception ( 5.3, 8.1, 8.3)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Tablets: 3 mg, 4 mg, and 5 mg. ( 3)
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
- Lactation: Advise not to breastfeed. ( 8.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflects exposure to BALVERSA as a single agent at the recommended dose (8 to 9 mg orally daily) in 479 patients with advanced urothelial cancer and FGFRalterations in 42756493BLC3001 (NCT03390504), 42756493BLC2001 (NCT02365597), 42756493BLC2002 (NCT 03473743), and 42756493EDI1001 (NCT01703481). Among 479 patients who received BALVERSA, the median duration of treatment was 4.8 months (range: 0.1 to 43 months). In this pooled safety population, the most common (>20%) adverse reactions, including laboratory abnormalities, were increased phosphate, nail disorders, stomatitis, diarrhea, increased creatinine, increased alkaline phosphatase, increased alanine aminotransferase, decreased hemoglobin, decreased sodium, increased aspartate aminotransferase, fatigue, dry mouth, dry skin, decreased phosphate, decreased appetite, dysgeusia, constipation, increased calcium, dry eye, palmar-plantar erythrodysesthesia syndrome, increased potassium, alopecia, and central serous retinopathy.
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA-approved patient labeling (Patient Information).
2.2 Recommended Dosage and Schedule
The recommended starting dose of BALVERSA is 8 mg (two 4 mg tablets) orally once daily, with a dose increase to 9 mg (three 3 mg tablets) once daily based on tolerability, including hyperphosphatemia, at 14 to 21 days [see Dosage and Administration (2.3)].
Swallow tablets whole with or without food. If vomiting occurs any time after taking BALVERSA, the next dose should be taken the next day. Treatment should continue until disease progression or unacceptable toxicity occurs.
If a dose of BALVERSA is missed, it can be taken as soon as possible on the same day. Resume the regular daily dose schedule for BALVERSA the next day. Extra tablets should not be taken to make up for the missed dose.
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
BALVERSA ®(erdafitinib) tablets are available in the strengths and packages listed below:
- 3 mg tablets: Yellow, round biconvex, film-coated, debossed with "3" on one side and "EF" on the other side.
- Bottle of 56-tablets with child resistant closure (NDC 59676-030-56).
- Bottle of 84-tablets with child resistant closure (NDC 59676-030-84).
- 4 mg tablets: Orange, round biconvex, film-coated, debossed with "4" on one side and "EF" on the other side.
- Bottle of 28-tablets with child resistant closure (NDC 59676-040-28).
- Bottle of 56-tablets with child resistant closure (NDC 59676-040-56).
- 5 mg tablets: Brown, round biconvex, film-coated, debossed with "5" on one side and "EF" on the other side.
- Bottle of 28-tablets with child resistant closure (NDC 59676-050-28).
7.1 Effect of Other Drugs On Balversa (7.1 Effect of Other Drugs on BALVERSA)
Table 7 summarizes drug interactions that affect the exposure of BALVERSA or serum phosphate level and their clinical management.
| Moderate CYP2C9 or Strong CYP3A4 Inhibitors | |
| Clinical Impact |
|
| Clinical Management |
|
| Strong CYP3A4 Inducers | |
| Clinical Impact |
|
| Clinical Management |
|
| Moderate CYP3A4 Inducers | |
| Clinical Impact |
|
| Clinical Management |
|
| Serum Phosphate Level-Altering Agents | |
| Clinical Impact |
|
| Clinical Management |
|
7.2 Effect of Balversa On Other Drugs (7.2 Effect of BALVERSA on Other Drugs)
Table 8 summarizes the effect of BALVERSA on other drugs and their clinical management.
| P-glycoprotein (P-gp) Substrates | |
|---|---|
| Clinical Impact |
|
| Clinical Management |
|
2.3 Dose Modifications for Adverse Reactions
The recommended dose modifications for adverse reactions are listed in Table 1.
| Dose | 1 stdose reduction | 2 nddose reduction | 3 rddose reduction | 4 thdose reduction | 5 thdose reduction |
|---|---|---|---|---|---|
|
9 mg ➞
(three 3 mg tablets) |
8 mg
(two 4 mg tablets) |
6 mg
(two 3 mg tablets) |
5 mg
(one 5 mg tablet) |
4 mg
(one 4 mg tablet) |
Stop |
|
8 mg ➞
(two 4 mg tablets) |
6 mg
(two 3 mg tablets) |
5 mg
(one 5 mg tablet) |
4 mg
(one 4 mg tablet) |
Stop |
Table 2 summarizes recommendations for dose interruption, reduction, or discontinuation of BALVERSA in the management of specific adverse reactions.
| Adverse Reaction | BALVERSA Dose Modification |
|---|---|
| Hyperphosphatemia | |
| In all patients, restrict phosphate intake to 600–800 mg daily. | |
| <6.99 mg/dL | Continue BALVERSA at current dose. |
| 7–8.99 mg/dL |
|
| 9–10 mg/dL |
|
| >10 mg/dL |
|
| Serum phosphate with life-threatening consequences; urgent intervention indicated (e.g., dialysis) |
|
| Central Serous Retinopathy (CSR) | |
| Any | Withhold BALVERSA and perform an ophthalmic evaluation within 2 weeks:
If recurs or has not improved after 4 weeks of withholding BALVERSA, consider permanent discontinuation. |
|
Other Adverse Reactions
Dose adjustment graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAEv5.0).
|
|
| Grade 3 | Withhold BALVERSA until resolves to Grade 1 or baseline, then may resume dose level lower. |
| Grade 4 | Permanently discontinue. |
8.3 Females and Males of Reproductive Potential
BALVERSA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)] .
14.1 Urothelial Carcinoma With Susceptible Fgfr3 (14.1 Urothelial Carcinoma with Susceptible FGFR3)
The efficacy of BALVERSA was evaluated in Study BLC3001 (NCT03390504) Cohort 1, a randomized, open-label, multicenter study in which 266 patients with advanced urothelial cancer harboring selected FGFR3alterations were randomized 1:1 to receive BALVERSA (8 mg with titration up to 9 mg) versus chemotherapy (docetaxel 75 mg/m 2once every 3 weeks or vinflunine 320 mg/m 2once every 3 weeks) until unacceptable toxicity or progression. Randomization was stratified by region (North America vs. Europe vs. rest of world), Eastern Cooperative Oncology Group (ECOG) performance status (0 or 1 vs. 2) and visceral or bone metastases (yes vs. no). All patients needed to have had disease progression after 1 or 2 prior treatments, at least 1 of which included a PD-1 or PD-L1 inhibitor. FGFR3genetic alterations were identified from tumor tissue in a central laboratory by the QIAGEN therascreen ® FGFRRGQ RT-Polymerase Chain Reaction (PCR) kit in 75% of patients while the remainder (25%) were identified by local next generation sequencing (NGS) assays.
The major efficacy outcome measures were overall survival (OS), progression-free survival (PFS), and objective response rate (ORR) assessed by investigator using RECIST (Response Evaluation Criteria in Solid Tumors) Version 1.1.
The median age was 67 years (range: 32 to 86 years) and 71% were male; 54% were White, 29% Asian, 0.4% Black, 0.4% multiple races, 16% not reported; 2% were Hispanic/Latino; and baseline ECOG performance status was 0 (43%), 1 (48%), or 2 (9%). Eighty-one percent of patients had FGFR3mutations, 17% had fusions, and 2% had both mutations and fusions. Ninety-five percent of patients had pure transitional cell carcinoma (TCC) and 5% had TCC with other histologic variants. The primary tumor location was the upper tract for 33% of subjects and lower tract for 67%; 74% of patients had visceral or bone metastases. Eighty-eight percent of patients received platinum-containing chemotherapy previously. PD-1 or PD-L1 inhibitor therapy was received only in the neoadjuvant or adjuvant setting in 7% of patients.
Statistically significant improvements in OS, PFS, and ORR were demonstrated for BALVERSA compared with chemotherapy.
Table 9 and Figures 1 and 2 summarize the efficacy results for BLC3001 Cohort 1.
| BALVERSA
N=136 |
Chemotherapy
N=130 |
|
|---|---|---|
| All p-values reported are 2-sided and compared with 0.019 of the allocated alpha for the interim analysis.
ORR = confirmed objective response (CR + PR) CI = Confidence Interval |
||
| Overall Survival (OS) | ||
| Number of events (%) | 77 (56.6%) | 78 (60.0%) |
| Median
Based on Kaplan-Meier estimates , months (95% CI)
|
12.1 (10.3, 16.4) | 7.8 (6.5, 11.1) |
| Hazard ratio
Based on an unstratified Cox proportional hazard model (95% CI)
|
0.64 (0.47, 0.88) | |
| p-value
Based on an unstratified log-rank test
|
0.0050 | |
| Progression-free survival (PFS) | ||
| Number of events (%) | 101 (74.3%) | 90 (69.2%) |
| Median , months (95% CI) | 5.6 (4.4, 5.7) | 2.7 (1.8, 3.7) |
| Hazard ratio (95% CI) | 0.58 (0.44, 0.78) | |
| p-value | 0.0002 | |
| Objective response rate (ORR) | ||
| ORR (95% CI) | 35.3% (27.3, 43.9) | 8.5% (4.3, 14.6) |
| p-value
p-value is estimated using Cochran-Haenszel (CMH) test with ECOG performance status (0 or 1 vs 2) as a stratification factor.
|
<0.001 | |
| Complete response, CR (%) | 5.1% | 0.8% |
| Partial response, PR (%) | 30.1% | 7.7% |
Figure 1: Kaplan-Meier Plot of Overall Survival (Study BLC3001 Cohort 1)
Figure 2: Kaplan-Meier Plot of Progression-free Survival (Study BLC3001 Cohort 1)
Principal Display Panel 3 Mg Tablet Bottle Carton (PRINCIPAL DISPLAY PANEL - 3 mg Tablet Bottle Carton)
NDC 59676-030-56
Balversa
®
(erdafitinib) tablets
3 mg
Each film-coated tablet
contains 3 mg of erdafitinib.
Rx only
56 film-coated tablets
Principal Display Panel 4 Mg Tablet Bottle Carton (PRINCIPAL DISPLAY PANEL - 4 mg Tablet Bottle Carton)
NDC 59676-040-56
Balversa
®
(erdafitinib) tablets
4 mg
Each film-coated tablet
contains 4 mg of erdafitinib.
Rx only
56 film-coated tablets
Principal Display Panel 5 Mg Tablet Bottle Carton (PRINCIPAL DISPLAY PANEL - 5 mg Tablet Bottle Carton)
NDC 59676-050-28
Balversa
®
(erdafitinib) tablets
5 mg
Each film-coated tablet
contains 5 mg of erdafitinib.
Rx only
28 film-coated tablets
5.2 Hyperphosphatemia and Soft Tissue Mineralization
BALVERSA can cause hyperphosphatemia leading to soft tissue mineralization, cutaneous calcinosis, non-uremic calciphylaxis and vascular calcification. Increases in phosphate levels are a pharmacodynamic effect of BALVERSA [see Pharmacodynamics (12.2)].
In the pooled safety population [see Adverse Reactions (6)], increased phosphate occurred in 73% of BALVERSA-treated patients. The median onset time of increased phosphate was 16 days (range: 8–421) after initiating BALVERSA. Twenty-four percent of patients received phosphate binders during treatment with BALVERSA. Vascular calcification was observed in 0.2% of patients treated with BALVERSA.
Monitor for hyperphosphatemia throughout treatment. Restrict dietary phosphate intake (600–800 mg daily) and avoid concomitant use of agents that may increase serum phosphate levels.
If serum phosphate is above 7.0 mg/dL, consider adding an oral phosphate binder until serum phosphate level returns to <7.0 mg/dL. Withhold, dose reduce, or permanently discontinue BALVERSA based on duration and severity of hyperphosphatemia according to Table 2 [see Dosage and Administration (2.3)].
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenicity studies have not been conducted with erdafitinib.
Erdafitinib was not mutagenic in a bacterial reverse mutation (Ames) assay and was not clastogenic in an in vitro micronucleus or an in vivo rat bone marrow micronucleus assay.
Fertility studies in animals have not been conducted with erdafitinib. In the 3-month repeat-dose toxicity study, erdafitinib showed effects on female reproductive organs (necrosis of the ovarian corpora lutea) in rats at an exposure less than the human exposure (AUC) at maximum recommended human dose.
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:43:43.981212 · Updated: 2026-03-14T22:48:06.394491