Polivy
20a16ab2-f338-4abb-9dcd-254bd949a2bc
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
POLIVY is a CD79b-directed antibody and microtubule inhibitor conjugate indicated: in combination with a rituximab product, cyclophosphamide, doxorubicin, and prednisone (R-CHP) for the treatment of adult patients who have previously untreated diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS) or high-grade B-cell lymphoma (HGBL) and who have an International Prognostic Index score of 2 or greater. ( 1.1 ) in combination with bendamustine and a rituximab product for the treatment of adult patients with relapsed or refractory DLBCL, NOS, after at least two prior therapies. ( 1.2 )
Dosage and Administration
The recommended dose of POLIVY is 1.8 mg/kg as an intravenous infusion every 21 days for 6 cycles. ( 2 ) Administer the initial POLIVY dose over 90 minutes. Subsequent infusions may be administered over 30 minutes if the previous infusion is tolerated. ( 2 ) Premedicate with an antihistamine and antipyretic before POLIVY. ( 2 ) See Full Prescribing Information for instructions on preparation and administration. ( 2.4 )
Contraindications
None.
Warnings and Precautions
Peripheral Neuropathy: Monitor patients for peripheral neuropathy and modify or discontinue dose accordingly. ( 5.1 ) Infusion-Related Reactions: Premedicate with an antihistamine and antipyretic. Monitor patients closely during infusions. Interrupt or discontinue infusion for reactions. ( 5.2 ) Myelosuppression: Monitor complete blood counts. Manage using dose delays or reductions and growth factor support. Monitor for signs of infection. ( 5.3 ) Serious and Opportunistic Infections: Closely monitor patients for signs of bacterial, fungal, or viral infections. ( 5.4 ) Progressive Multifocal Leukoencephalopathy (PML): Monitor patients for new or worsening neurological, cognitive, or behavioral changes suggestive of PML. ( 5.5 ) Tumor Lysis Syndrome: Closely monitor patients with high tumor burden or rapidly proliferative tumors. ( 5.6 ) Hepatotoxicity: Monitor liver enzymes and bilirubin. ( 5.7 ) Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for 3 months after the last dose. ( 5.8 )
Adverse Reactions
The following clinically significant adverse reactions are discussed in greater detail in other sections of the label: Peripheral Neuropathy [see Warnings and Precautions (5.1) ] Infusion-Related Reactions [see Warnings and Precautions (5.2) ] Myelosuppression [see Warnings and Precautions (5.3) ] Serious and Opportunistic Infections [see Warnings and Precautions (5.4) ] Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.5) ] Tumor Lysis Syndrome [see Warnings and Precautions (5.6) ] Hepatotoxicity [see Warnings and Precautions (5.7) ]
Drug Interactions
Concomitant use of strong CYP3A inhibitors or inducers has the potential to affect the exposure to unconjugated monomethyl auristatin E (MMAE). ( 7.1 )
Description
Indications and Usage ( 1.1 , 1.2 ) 4/2023 Dosage and Administration ( 2.1 . 2.2 , 2.3 , 2.4 ) 4/2023 Warnings and Precautions ( 5.1 , 5.2 . 5.3 , 5.4 , 5.7 ) 4/2023
Medication Information
Warnings and Precautions
Peripheral Neuropathy: Monitor patients for peripheral neuropathy and modify or discontinue dose accordingly. ( 5.1 ) Infusion-Related Reactions: Premedicate with an antihistamine and antipyretic. Monitor patients closely during infusions. Interrupt or discontinue infusion for reactions. ( 5.2 ) Myelosuppression: Monitor complete blood counts. Manage using dose delays or reductions and growth factor support. Monitor for signs of infection. ( 5.3 ) Serious and Opportunistic Infections: Closely monitor patients for signs of bacterial, fungal, or viral infections. ( 5.4 ) Progressive Multifocal Leukoencephalopathy (PML): Monitor patients for new or worsening neurological, cognitive, or behavioral changes suggestive of PML. ( 5.5 ) Tumor Lysis Syndrome: Closely monitor patients with high tumor burden or rapidly proliferative tumors. ( 5.6 ) Hepatotoxicity: Monitor liver enzymes and bilirubin. ( 5.7 ) Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for 3 months after the last dose. ( 5.8 )
Indications and Usage
POLIVY is a CD79b-directed antibody and microtubule inhibitor conjugate indicated: in combination with a rituximab product, cyclophosphamide, doxorubicin, and prednisone (R-CHP) for the treatment of adult patients who have previously untreated diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS) or high-grade B-cell lymphoma (HGBL) and who have an International Prognostic Index score of 2 or greater. ( 1.1 ) in combination with bendamustine and a rituximab product for the treatment of adult patients with relapsed or refractory DLBCL, NOS, after at least two prior therapies. ( 1.2 )
Dosage and Administration
The recommended dose of POLIVY is 1.8 mg/kg as an intravenous infusion every 21 days for 6 cycles. ( 2 ) Administer the initial POLIVY dose over 90 minutes. Subsequent infusions may be administered over 30 minutes if the previous infusion is tolerated. ( 2 ) Premedicate with an antihistamine and antipyretic before POLIVY. ( 2 ) See Full Prescribing Information for instructions on preparation and administration. ( 2.4 )
Contraindications
None.
Adverse Reactions
The following clinically significant adverse reactions are discussed in greater detail in other sections of the label: Peripheral Neuropathy [see Warnings and Precautions (5.1) ] Infusion-Related Reactions [see Warnings and Precautions (5.2) ] Myelosuppression [see Warnings and Precautions (5.3) ] Serious and Opportunistic Infections [see Warnings and Precautions (5.4) ] Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.5) ] Tumor Lysis Syndrome [see Warnings and Precautions (5.6) ] Hepatotoxicity [see Warnings and Precautions (5.7) ]
Drug Interactions
Concomitant use of strong CYP3A inhibitors or inducers has the potential to affect the exposure to unconjugated monomethyl auristatin E (MMAE). ( 7.1 )
Description
Indications and Usage ( 1.1 , 1.2 ) 4/2023 Dosage and Administration ( 2.1 . 2.2 , 2.3 , 2.4 ) 4/2023 Warnings and Precautions ( 5.1 , 5.2 . 5.3 , 5.4 , 5.7 ) 4/2023
Section 42229-5
Patients with Previously Untreated DLBCL, NOS or HGBL
The recommended dosage of POLIVY is 1.8 mg/kg administered as an intravenous infusion every 21 days for 6 cycles in combination with a rituximab product, cyclophosphamide, doxorubicin, and prednisone [see Clinical Studies (14.1)]. Administer POLIVY, cyclophosphamide, doxorubicin, and a rituximab product in any order on Day 1 after the administration of prednisone. Prednisone is administered on Days 1–5 of each cycle.
Section 43683-2
Section 44425-7
Storage and Handling
Store refrigerated at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not use beyond the expiration date shown on the carton. Do not freeze. Do not shake.
POLIVY is a hazardous drug. Follow applicable special handling and disposal procedures.1
15 References
- "OSHA Hazardous Drugs." OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
11 Description
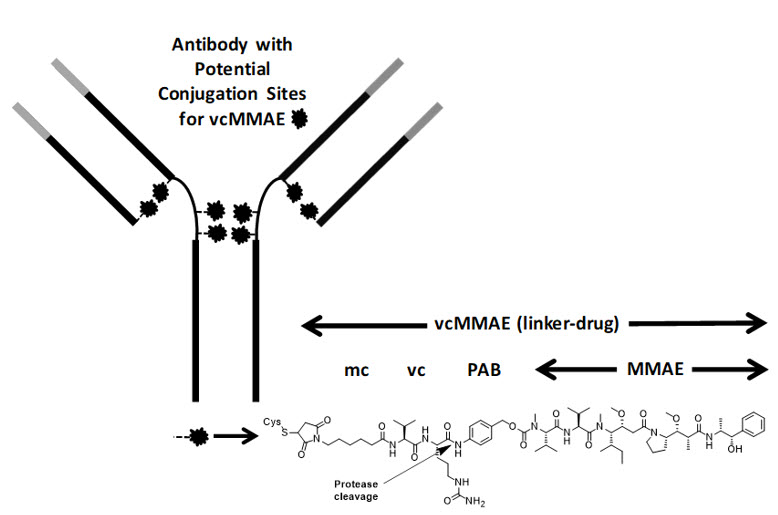
Polatuzumab vedotin-piiq is a CD79b-directed antibody and microtubule inhibitor conjugate. It consists of three components: 1) the humanized immunoglobulin G1 (IgG1) monoclonal antibody specific for human CD79b; 2) the small molecule anti-mitotic agent MMAE; and 3) a protease-cleavable linker maleimidocaproyl-valine-citrulline-p-aminobenzyloxycarbonyl (mc-vc-PAB) that covalently attaches MMAE to the polatuzumab antibody.
Polatuzumab vedotin-piiq has an approximate molecular weight of 150 kDa. An average of 3.5 molecules of MMAE are attached to each antibody molecule. Polatuzumab vedotin-piiq is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
POLIVY (polatuzumab vedotin-piiq) for injection is supplied as a sterile, white to grayish-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution.
Each single-dose 30 mg POLIVY vial delivers 30 mg of polatuzumab vedotin-piiq, polysorbate-20 (1.8 mg), sodium hydroxide (0.82 mg), succinic acid (1.77 mg), and sucrose (62 mg). After reconstitution with 1.8 mL of Sterile Water for Injection, USP, the final concentration is 20 mg/mL with a pH of approximately 5.3.
Each single-dose 140 mg POLIVY vial delivers 140 mg of polatuzumab vedotin-piiq, polysorbate-20 (8.4 mg), sodium hydroxide (3.80 mg), succinic acid (8.27 mg), and sucrose (288 mg). After reconstitution with 7.2 mL of Sterile Water for Injection, USP, the final concentration is 20 mg/mL with a pH of approximately 5.3.
The POLIVY vial stoppers are not made with natural rubber latex.
8.4 Pediatric Use
Safety and effectiveness of POLIVY have not been established in pediatric patients.
8.5 Geriatric Use
Among 435 patients treated with POLIVY plus R-CHP in POLARIX, 227 (52%) were ≥65 years of age. No overall differences in safety or efficacy were observed between patients aged ≥65 years and younger patients.
Among 173 patients treated with POLIVY plus BR in Study GO29365, 95 (55%) were ≥65 years of age. Patients aged ≥65 had a numerically higher incidence of serious adverse reactions (64%) than patients aged <65 (53%). This study did not include sufficient numbers of patients to determine whether efficacy differed in patients aged ≥65 and younger patients.
5.7 Hepatotoxicity
Serious cases of hepatotoxicity that were consistent with hepatocellular injury, including elevations of transaminases and/or bilirubin, have occurred in patients treated with POLIVY.
In recipients of POLIVY plus R-CHP, Grade 3–4 elevation of ALT and AST developed in 1.4% and 0.7% of patients, respectively.
In Study GO29365, Grade 3 and Grade 4 transaminase elevations each developed in 1.9% of patients.
Preexisting liver disease, elevated baseline liver enzymes, and concomitant medications may increase the risk of hepatotoxicity. Monitor liver enzymes and bilirubin level.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of POLIVY or of other polatuzumab products.
In studies POLARIX and GO29365, 1.4% (6/427) and 6% (8/134) of patients tested positive for antibodies against polatuzumab vedotin-piiq, respectively, of which none were positive for neutralizing antibodies. Because of the low occurrence of anti-drug antibodies, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of polatuzumab products is unknown.
4 Contraindications
None.
6 Adverse Reactions
The following clinically significant adverse reactions are discussed in greater detail in other sections of the label:
- Peripheral Neuropathy [see Warnings and Precautions (5.1)]
- Infusion-Related Reactions [see Warnings and Precautions (5.2)]
- Myelosuppression [see Warnings and Precautions (5.3)]
- Serious and Opportunistic Infections [see Warnings and Precautions (5.4)]
- Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.5)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.6)]
- Hepatotoxicity [see Warnings and Precautions (5.7)]
7 Drug Interactions
Concomitant use of strong CYP3A inhibitors or inducers has the potential to affect the exposure to unconjugated monomethyl auristatin E (MMAE). (7.1)
5.3 Myelosuppression
Treatment with POLIVY can cause serious or severe myelosuppression, including neutropenia, thrombocytopenia, and anemia.
In POLARIX, 90% of patients treated with POLIVY plus R-CHP had primary prophylaxis with G-CSF. Grade 3–4 hematologic adverse reactions included lymphopenia (44%), neutropenia (39%), febrile neutropenia (15%), anemia (14%), and thrombocytopenia (8%) [see Adverse Reactions (6.1)].
In Study GO29365, in patients treated with POLIVY plus BR (n = 45), 42% received primary prophylaxis with G-CSF. Grade 3 or higher hematologic adverse reactions included neutropenia (42%), thrombocytopenia (40%), anemia (24%), lymphopenia (13%), and febrile neutropenia (11%) [see Adverse Reactions (6.1)]. Grade 4 hematologic adverse reactions included neutropenia (24%), thrombocytopenia (16%), lymphopenia (9%), and febrile neutropenia (4.4%). Cytopenias were the most common reason for treatment discontinuation (18% of all patients).
Monitor complete blood counts throughout treatment. Cytopenias may require a delay, dose reduction, or discontinuation of POLIVY [see Dosage and Administration (2.2)]. Administer prophylactic G-CSF for neutropenia in patients receiving POLIVY plus R-CHP. Consider prophylactic G-CSF administration in patients receiving POLIVY plus bendamustine and a rituximab product.
12.2 Pharmacodynamics
Over polatuzumab vedotin-piiq dosages of 0.1 to 2.4 mg/kg (0.06 to 1.33 times the approved recommended dosage), higher exposures (AUC and Cmax) of antibody-conjugated MMAE (acMMAE) and unconjugated MMAE were associated with higher incidence of some adverse reactions (including ≥Grade 3 thrombocytopenia and ≥Grade 3 anemia). Higher exposure (AUC and Cmax) of unconjugated MMAE was associated with higher incidence of ≥Grade 2 peripheral neuropathy. Lower acMMAE exposure (AUC) was associated with lower efficacy in patients with relapsed or refractory DLBCL.
12.3 Pharmacokinetics
The exposure parameters of acMMAE and unconjugated MMAE (the cytotoxic component of polatuzumab vedotin-piiq) are summarized in Table 10. The plasma exposure of acMMAE and unconjugated MMAE increased proportionally over a polatuzumab vedotin-piiq dose range from 0.1 to 2.4 mg/kg (0.06 to 1.33 times the approved recommended dosage). Cycle 3 acMMAE AUC were predicted to increase by approximately 30% over Cycle 1 AUC, and achieved more than 90% of the Cycle 6 AUC. Unconjugated MMAE plasma exposures were <3% of acMMAE exposures, and the AUC and Cmax were predicted to decrease after repeated every-3-week dosing.
| R/R DLBCL, NOS | Previously Untreated DLBCL, NOS or HGBL | |||
|---|---|---|---|---|
| acMMAE | Unconjugated MMAE | acMMAE | Unconjugated MMAE | |
| Cmax=maximum concentration; AUC=area under the concentration-time curve from time zero to day 21. | ||||
| Cmax (ng/mL) | 688 (15%) | 3.19 (57%) | 587 (15%) | 2.45 (46%) |
| AUC (day*ng/mL) | 2040 (35%) | 31.0 (56%) | 1690 (22%) | 20.8 (50%) |
8.6 Hepatic Impairment
Avoid the administration of POLIVY in patients with moderate or severe hepatic impairment (total bilirubin greater than 1.5 × ULN and any AST). Patients with moderate or severe hepatic impairment are likely to have increased exposure to MMAE, which may increase the risk of adverse reactions. POLIVY has not been studied in patients with moderate or severe hepatic impairment [see Clinical Pharmacology (12.3) and Warnings and Precautions (5.7)].
No adjustment in the starting dose is required when administering POLIVY to patients with mild hepatic impairment (total bilirubin 1 to 1.5 × ULN or AST greater than ULN).
1 Indications and Usage
POLIVY is a CD79b-directed antibody and microtubule inhibitor conjugate indicated:
- in combination with a rituximab product, cyclophosphamide, doxorubicin, and prednisone (R-CHP) for the treatment of adult patients who have previously untreated diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS) or high-grade B-cell lymphoma (HGBL) and who have an International Prognostic Index score of 2 or greater. (1.1)
- in combination with bendamustine and a rituximab product for the treatment of adult patients with relapsed or refractory DLBCL, NOS, after at least two prior therapies. (1.2)
12.1 Mechanism of Action
Polatuzumab vedotin-piiq is a CD79b-directed antibody-drug conjugate with activity against dividing B cells. The small molecule, MMAE, is an anti-mitotic agent covalently attached to the antibody via a cleavable linker. The monoclonal antibody binds to CD79b, a B-cell specific surface protein, which is a component of the B-cell receptor. Upon binding CD79b, polatuzumab vedotin-piiq is internalized, and the linker is cleaved by lysosomal proteases to enable intracellular delivery of MMAE. MMAE binds to microtubules and kills dividing cells by inhibiting cell division and inducing apoptosis.
5.6 Tumor Lysis Syndrome
POLIVY may cause tumor lysis syndrome. Patients with high tumor burden and rapidly proliferative tumor may be at increased risk of tumor lysis syndrome. Monitor closely and take appropriate measures, including tumor lysis syndrome prophylaxis.
5.1 Peripheral Neuropathy
POLIVY can cause peripheral neuropathy, including severe cases. Peripheral neuropathy occurs as early as the first cycle of treatment and is a cumulative effect [see Adverse Reactions (6.1)]. POLIVY may exacerbate pre-existing peripheral neuropathy.
In POLARIX, of 435 patients treated with POLIVY plus R-CHP, 53% reported new or worsening peripheral neuropathy, with a median time to onset of 2.3 months. Peripheral neuropathy was Grade 1 in 39% of patients, Grade 2 in 12%, and Grade 3 in 1.6%. Peripheral neuropathy resulted in dose reduction in 4% of treated patients and treatment discontinuation in 0.7%. Among patients with peripheral neuropathy after POLIVY, 58% reported resolution after a median of 4 months.
In Study GO29365, of 173 patients treated with POLIVY, 40% reported new or worsening peripheral neuropathy, with a median time to onset of 2.1 months. Peripheral neuropathy was Grade 1 in 26% of patients, Grade 2 in 12%, and Grade 3 in 2.3%. Peripheral neuropathy resulted in POLIVY dose reduction in 2.9% of treated patients, dose delay in 1.2%, and permanent discontinuation in 2.9%. Sixty-five percent of patients reported improvement or resolution of peripheral neuropathy after a median of 1 month, and 48% reported complete resolution.
The peripheral neuropathy is predominantly sensory; however, motor and sensorimotor peripheral neuropathy also occur. Monitor for symptoms of peripheral neuropathy such as hypoesthesia, hyperesthesia, paresthesia, dysesthesia, neuropathic pain, burning sensation, weakness, or gait disturbance. Patients experiencing new or worsening peripheral neuropathy may require a delay, dose reduction, or discontinuation of POLIVY [see Dosage and Administration (2.2)].
5.8 Embryo Fetal Toxicity
Based on the mechanism of action and findings from animal studies, POLIVY can cause fetal harm when administered to a pregnant woman. The small molecule component of POLIVY, MMAE, administered to rats caused adverse developmental outcomes, including embryo-fetal mortality and structural abnormalities, at exposures below those occurring clinically at the recommended dose.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with POLIVY and for 3 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with POLIVY and for 5 months after the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
5 Warnings and Precautions
- Peripheral Neuropathy: Monitor patients for peripheral neuropathy and modify or discontinue dose accordingly. (5.1)
- Infusion-Related Reactions: Premedicate with an antihistamine and antipyretic. Monitor patients closely during infusions. Interrupt or discontinue infusion for reactions. (5.2)
- Myelosuppression: Monitor complete blood counts. Manage using dose delays or reductions and growth factor support. Monitor for signs of infection. (5.3)
- Serious and Opportunistic Infections: Closely monitor patients for signs of bacterial, fungal, or viral infections. (5.4)
- Progressive Multifocal Leukoencephalopathy (PML): Monitor patients for new or worsening neurological, cognitive, or behavioral changes suggestive of PML. (5.5)
- Tumor Lysis Syndrome: Closely monitor patients with high tumor burden or rapidly proliferative tumors. (5.6)
- Hepatotoxicity: Monitor liver enzymes and bilirubin. (5.7)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for 3 months after the last dose. (5.8)
2 Dosage and Administration
- The recommended dose of POLIVY is 1.8 mg/kg as an intravenous infusion every 21 days for 6 cycles. (2)
- Administer the initial POLIVY dose over 90 minutes. Subsequent infusions may be administered over 30 minutes if the previous infusion is tolerated. (2)
- Premedicate with an antihistamine and antipyretic before POLIVY. (2)
- See Full Prescribing Information for instructions on preparation and administration. (2.4)
3 Dosage Forms and Strengths
For injection: 30 mg/vial or 140 mg/vial of polatuzumab vedotin-piiq as a white to grayish-white lyophilized powder in a single-dose vial for reconstitution and further dilution.
8 Use in Specific Populations
5.2 Infusion Related Reactions
POLIVY can cause infusion-related reactions, including severe cases. Delayed infusion-related reactions as late as 24 hours after receiving POLIVY have occurred.
With premedication, 13% of patients (58/435) in POLARIX reported infusion-related reactions after the administration of POLIVY plus R-CHP. The reactions were Grade 1 in 4.4% of patients, Grade 2 in 8%, and Grade 3 in 1.1%.
With premedication, 7% of patients (12/173) in Study GO29365 reported infusion-related reactions after the administration of POLIVY. The reactions were Grade 1 in 4.6% of patients, Grade 2 in 1.7%, and Grade 3 in 0.6%.
Symptoms occurring in ≥1% of patients included chills, dyspnea, pyrexia, pruritus, rash, and chest discomfort. Administer an antihistamine and antipyretic prior to the administration of POLIVY, and monitor patients closely throughout the infusion. If an infusion-related reaction occurs, interrupt the infusion and institute appropriate medical management [see Dosage and Administration (2.2)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described below reflect exposure to POLIVY 1.8 mg/kg in 480 patients with large B-cell lymphoma (LBCL), including those with previously untreated LBCL (POLARIX) and relapsed or refractory DLBCL (GO29365).
1.2 Relapsed Or Refractory Dlbcl, Nos
POLIVY in combination with bendamustine and a rituximab product is indicated for the treatment of adult patients with relapsed or refractory DLBCL, NOS, after at least two prior therapies.
2.3 Recommended Prophylactic Medications
If not already premedicated for a rituximab product, administer an antihistamine and antipyretic at least 30 to 60 minutes prior to POLIVY for potential infusion-related reactions [see Warnings and Precautions (5.2)].
Administer prophylaxis for Pneumocystis jiroveci pneumonia and herpesvirus throughout treatment with POLIVY.
Administer prophylactic granulocyte colony-stimulating factor (G-CSF) for neutropenia in patients receiving POLIVY plus R-CHP. Consider prophylactic G-CSF administration for neutropenia in patients receiving POLIVY plus bendamustine and a rituximab product [see Warnings and Precautions (5.3)].
Administer tumor lysis syndrome prophylaxis for patients at increased risk of tumor lysis syndrome [see Warnings and Precautions (5.6)].
5.4 Serious and Opportunistic Infections
Fatal and/or serious infections, including opportunistic infections such as sepsis, pneumonia (including Pneumocystis jiroveci and other fungal pneumonia), herpesvirus infection, and cytomegalovirus infection have occurred in patients treated with POLIVY [see Adverse Reactions (6.1)].
In POLARIX, Grade 3–4 infections occurred in 14% (61/435) of patients treated with POLIVY plus R-CHP and infection-related deaths were reported in 1.1% of patients.
In Study GO29365, Grade 3 or higher infections occurred in 32% (55/173) of patients treated with POLIVYand infection-related deaths were reported in 2.9% of patients within 90 days of last treatment.
Closely monitor patients during treatment for signs of infection. Administer prophylaxis for Pneumocystis jiroveci pneumonia and herpesvirus. Administer prophylactic G-CSF for neutropenia as recommended [see Dosage and Administration (2.3)].
1.1 Previously Untreated Dlbcl, Nos Or Hgbl
POLIVY in combination with a rituximab product, cyclophosphamide, doxorubicin, and prednisone (R-CHP) is indicated for the treatment of adult patients who have previously untreated diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS) or high-grade B-cell lymphoma (HGBL) and who have an International Prognostic Index score of 2 or greater.
Principal Display Panel 30 Mg Vial Carton
NDC 50242-103-01
Polivy®
(polatuzumab vedotin-piiq)
For Injection
30 mg per vial
For Intravenous Infusion Only.
Reconstitute and Dilute
prior to administration.
Single-Dose Vial.
Discard unused portion.
CAUTION: Hazardous Agent
Rx only
1 vial
Genentech
11008718
Principal Display Panel 140 Mg Vial Carton
NDC 50242-105-01
Polivy®
(polatuzumab vedotin-piiq)
For Injection
140 mg per vial
For Intravenous Infusion Only.
Reconstitute and Dilute
prior to administration.
Single-Dose Vial.
Discard unused portion.
CAUTION: Hazardous Agent
Rx only
1 vial
Genentech
11007928
8.3 Females and Males of Reproductive Potential
POLIVY can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
2.4 Instructions for Preparation and Administration
Reconstitute and further dilute POLIVY prior to intravenous infusion.
POLIVY is a hazardous drug. Follow applicable special handling and disposal procedures.1
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
5.5 Progressive Multifocal Leukoencephalopathy (pml)
PML has been reported after treatment with POLIVY plus bendamustine and obinutuzumab in study GO29365 (0.6%, 1/173). Monitor for new or worsening neurological, cognitive, or behavioral changes. Hold POLIVY and any concomitant chemotherapy if PML is suspected, and permanently discontinue if the diagnosis is confirmed.
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies in animals have not been performed with polatuzumab vedotin-piiq or MMAE.
MMAE was positive for genotoxicity in the in vivo rat bone marrow micronucleus study through an aneugenic mechanism. MMAE was not mutagenic in the bacterial reverse mutation (Ames) assay or the L5178Y mouse lymphoma forward mutation assay.
Fertility studies in animals have not been performed with polatuzumab vedotin-piiq or MMAE. However, results of repeat-dose toxicity indicate the potential for polatuzumab vedotin-piiq to impair female and male fertility. In the 4-week repeat-dose toxicity study in rats with weekly dosing of 2, 6, and 10 mg/kg, dose-dependent testicular seminiferous tubule degeneration with abnormal lumen contents in the epididymis was observed. Findings in the testes and epididymis did not reverse and correlated with decreased testes weight and gross findings of small and/or soft testes at recovery necropsy in males given doses ≥2 mg/kg (below the exposure at the recommended dose based on unconjugated MMAE AUC).
MMAE-containing ADCs have been associated with adverse ovarian effects when administered to sexually immature animals. Adverse effects included decrease in, or absence of, secondary and tertiary ovarian follicles after weekly administration to cynomolgus monkeys in studies of 4-week duration. These effects showed a trend towards recovery 6 weeks after the end of dosing; no changes were observed in primordial follicles.
Structured Label Content
Section 42229-5 (42229-5)
Patients with Previously Untreated DLBCL, NOS or HGBL
The recommended dosage of POLIVY is 1.8 mg/kg administered as an intravenous infusion every 21 days for 6 cycles in combination with a rituximab product, cyclophosphamide, doxorubicin, and prednisone [see Clinical Studies (14.1)]. Administer POLIVY, cyclophosphamide, doxorubicin, and a rituximab product in any order on Day 1 after the administration of prednisone. Prednisone is administered on Days 1–5 of each cycle.
Section 43683-2 (43683-2)
Section 44425-7 (44425-7)
Storage and Handling
Store refrigerated at 2°C to 8°C (36°F to 46°F) in original carton to protect from light. Do not use beyond the expiration date shown on the carton. Do not freeze. Do not shake.
POLIVY is a hazardous drug. Follow applicable special handling and disposal procedures.1
15 References (15 REFERENCES)
- "OSHA Hazardous Drugs." OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
11 Description (11 DESCRIPTION)
Polatuzumab vedotin-piiq is a CD79b-directed antibody and microtubule inhibitor conjugate. It consists of three components: 1) the humanized immunoglobulin G1 (IgG1) monoclonal antibody specific for human CD79b; 2) the small molecule anti-mitotic agent MMAE; and 3) a protease-cleavable linker maleimidocaproyl-valine-citrulline-p-aminobenzyloxycarbonyl (mc-vc-PAB) that covalently attaches MMAE to the polatuzumab antibody.
Polatuzumab vedotin-piiq has an approximate molecular weight of 150 kDa. An average of 3.5 molecules of MMAE are attached to each antibody molecule. Polatuzumab vedotin-piiq is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
POLIVY (polatuzumab vedotin-piiq) for injection is supplied as a sterile, white to grayish-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution.
Each single-dose 30 mg POLIVY vial delivers 30 mg of polatuzumab vedotin-piiq, polysorbate-20 (1.8 mg), sodium hydroxide (0.82 mg), succinic acid (1.77 mg), and sucrose (62 mg). After reconstitution with 1.8 mL of Sterile Water for Injection, USP, the final concentration is 20 mg/mL with a pH of approximately 5.3.
Each single-dose 140 mg POLIVY vial delivers 140 mg of polatuzumab vedotin-piiq, polysorbate-20 (8.4 mg), sodium hydroxide (3.80 mg), succinic acid (8.27 mg), and sucrose (288 mg). After reconstitution with 7.2 mL of Sterile Water for Injection, USP, the final concentration is 20 mg/mL with a pH of approximately 5.3.
The POLIVY vial stoppers are not made with natural rubber latex.
8.4 Pediatric Use
Safety and effectiveness of POLIVY have not been established in pediatric patients.
8.5 Geriatric Use
Among 435 patients treated with POLIVY plus R-CHP in POLARIX, 227 (52%) were ≥65 years of age. No overall differences in safety or efficacy were observed between patients aged ≥65 years and younger patients.
Among 173 patients treated with POLIVY plus BR in Study GO29365, 95 (55%) were ≥65 years of age. Patients aged ≥65 had a numerically higher incidence of serious adverse reactions (64%) than patients aged <65 (53%). This study did not include sufficient numbers of patients to determine whether efficacy differed in patients aged ≥65 and younger patients.
5.7 Hepatotoxicity
Serious cases of hepatotoxicity that were consistent with hepatocellular injury, including elevations of transaminases and/or bilirubin, have occurred in patients treated with POLIVY.
In recipients of POLIVY plus R-CHP, Grade 3–4 elevation of ALT and AST developed in 1.4% and 0.7% of patients, respectively.
In Study GO29365, Grade 3 and Grade 4 transaminase elevations each developed in 1.9% of patients.
Preexisting liver disease, elevated baseline liver enzymes, and concomitant medications may increase the risk of hepatotoxicity. Monitor liver enzymes and bilirubin level.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of POLIVY or of other polatuzumab products.
In studies POLARIX and GO29365, 1.4% (6/427) and 6% (8/134) of patients tested positive for antibodies against polatuzumab vedotin-piiq, respectively, of which none were positive for neutralizing antibodies. Because of the low occurrence of anti-drug antibodies, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of polatuzumab products is unknown.
4 Contraindications (4 CONTRAINDICATIONS)
None.
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following clinically significant adverse reactions are discussed in greater detail in other sections of the label:
- Peripheral Neuropathy [see Warnings and Precautions (5.1)]
- Infusion-Related Reactions [see Warnings and Precautions (5.2)]
- Myelosuppression [see Warnings and Precautions (5.3)]
- Serious and Opportunistic Infections [see Warnings and Precautions (5.4)]
- Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.5)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.6)]
- Hepatotoxicity [see Warnings and Precautions (5.7)]
7 Drug Interactions (7 DRUG INTERACTIONS)
Concomitant use of strong CYP3A inhibitors or inducers has the potential to affect the exposure to unconjugated monomethyl auristatin E (MMAE). (7.1)
5.3 Myelosuppression
Treatment with POLIVY can cause serious or severe myelosuppression, including neutropenia, thrombocytopenia, and anemia.
In POLARIX, 90% of patients treated with POLIVY plus R-CHP had primary prophylaxis with G-CSF. Grade 3–4 hematologic adverse reactions included lymphopenia (44%), neutropenia (39%), febrile neutropenia (15%), anemia (14%), and thrombocytopenia (8%) [see Adverse Reactions (6.1)].
In Study GO29365, in patients treated with POLIVY plus BR (n = 45), 42% received primary prophylaxis with G-CSF. Grade 3 or higher hematologic adverse reactions included neutropenia (42%), thrombocytopenia (40%), anemia (24%), lymphopenia (13%), and febrile neutropenia (11%) [see Adverse Reactions (6.1)]. Grade 4 hematologic adverse reactions included neutropenia (24%), thrombocytopenia (16%), lymphopenia (9%), and febrile neutropenia (4.4%). Cytopenias were the most common reason for treatment discontinuation (18% of all patients).
Monitor complete blood counts throughout treatment. Cytopenias may require a delay, dose reduction, or discontinuation of POLIVY [see Dosage and Administration (2.2)]. Administer prophylactic G-CSF for neutropenia in patients receiving POLIVY plus R-CHP. Consider prophylactic G-CSF administration in patients receiving POLIVY plus bendamustine and a rituximab product.
12.2 Pharmacodynamics
Over polatuzumab vedotin-piiq dosages of 0.1 to 2.4 mg/kg (0.06 to 1.33 times the approved recommended dosage), higher exposures (AUC and Cmax) of antibody-conjugated MMAE (acMMAE) and unconjugated MMAE were associated with higher incidence of some adverse reactions (including ≥Grade 3 thrombocytopenia and ≥Grade 3 anemia). Higher exposure (AUC and Cmax) of unconjugated MMAE was associated with higher incidence of ≥Grade 2 peripheral neuropathy. Lower acMMAE exposure (AUC) was associated with lower efficacy in patients with relapsed or refractory DLBCL.
12.3 Pharmacokinetics
The exposure parameters of acMMAE and unconjugated MMAE (the cytotoxic component of polatuzumab vedotin-piiq) are summarized in Table 10. The plasma exposure of acMMAE and unconjugated MMAE increased proportionally over a polatuzumab vedotin-piiq dose range from 0.1 to 2.4 mg/kg (0.06 to 1.33 times the approved recommended dosage). Cycle 3 acMMAE AUC were predicted to increase by approximately 30% over Cycle 1 AUC, and achieved more than 90% of the Cycle 6 AUC. Unconjugated MMAE plasma exposures were <3% of acMMAE exposures, and the AUC and Cmax were predicted to decrease after repeated every-3-week dosing.
| R/R DLBCL, NOS | Previously Untreated DLBCL, NOS or HGBL | |||
|---|---|---|---|---|
| acMMAE | Unconjugated MMAE | acMMAE | Unconjugated MMAE | |
| Cmax=maximum concentration; AUC=area under the concentration-time curve from time zero to day 21. | ||||
| Cmax (ng/mL) | 688 (15%) | 3.19 (57%) | 587 (15%) | 2.45 (46%) |
| AUC (day*ng/mL) | 2040 (35%) | 31.0 (56%) | 1690 (22%) | 20.8 (50%) |
8.6 Hepatic Impairment
Avoid the administration of POLIVY in patients with moderate or severe hepatic impairment (total bilirubin greater than 1.5 × ULN and any AST). Patients with moderate or severe hepatic impairment are likely to have increased exposure to MMAE, which may increase the risk of adverse reactions. POLIVY has not been studied in patients with moderate or severe hepatic impairment [see Clinical Pharmacology (12.3) and Warnings and Precautions (5.7)].
No adjustment in the starting dose is required when administering POLIVY to patients with mild hepatic impairment (total bilirubin 1 to 1.5 × ULN or AST greater than ULN).
1 Indications and Usage (1 INDICATIONS AND USAGE)
POLIVY is a CD79b-directed antibody and microtubule inhibitor conjugate indicated:
- in combination with a rituximab product, cyclophosphamide, doxorubicin, and prednisone (R-CHP) for the treatment of adult patients who have previously untreated diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS) or high-grade B-cell lymphoma (HGBL) and who have an International Prognostic Index score of 2 or greater. (1.1)
- in combination with bendamustine and a rituximab product for the treatment of adult patients with relapsed or refractory DLBCL, NOS, after at least two prior therapies. (1.2)
12.1 Mechanism of Action
Polatuzumab vedotin-piiq is a CD79b-directed antibody-drug conjugate with activity against dividing B cells. The small molecule, MMAE, is an anti-mitotic agent covalently attached to the antibody via a cleavable linker. The monoclonal antibody binds to CD79b, a B-cell specific surface protein, which is a component of the B-cell receptor. Upon binding CD79b, polatuzumab vedotin-piiq is internalized, and the linker is cleaved by lysosomal proteases to enable intracellular delivery of MMAE. MMAE binds to microtubules and kills dividing cells by inhibiting cell division and inducing apoptosis.
5.6 Tumor Lysis Syndrome
POLIVY may cause tumor lysis syndrome. Patients with high tumor burden and rapidly proliferative tumor may be at increased risk of tumor lysis syndrome. Monitor closely and take appropriate measures, including tumor lysis syndrome prophylaxis.
5.1 Peripheral Neuropathy
POLIVY can cause peripheral neuropathy, including severe cases. Peripheral neuropathy occurs as early as the first cycle of treatment and is a cumulative effect [see Adverse Reactions (6.1)]. POLIVY may exacerbate pre-existing peripheral neuropathy.
In POLARIX, of 435 patients treated with POLIVY plus R-CHP, 53% reported new or worsening peripheral neuropathy, with a median time to onset of 2.3 months. Peripheral neuropathy was Grade 1 in 39% of patients, Grade 2 in 12%, and Grade 3 in 1.6%. Peripheral neuropathy resulted in dose reduction in 4% of treated patients and treatment discontinuation in 0.7%. Among patients with peripheral neuropathy after POLIVY, 58% reported resolution after a median of 4 months.
In Study GO29365, of 173 patients treated with POLIVY, 40% reported new or worsening peripheral neuropathy, with a median time to onset of 2.1 months. Peripheral neuropathy was Grade 1 in 26% of patients, Grade 2 in 12%, and Grade 3 in 2.3%. Peripheral neuropathy resulted in POLIVY dose reduction in 2.9% of treated patients, dose delay in 1.2%, and permanent discontinuation in 2.9%. Sixty-five percent of patients reported improvement or resolution of peripheral neuropathy after a median of 1 month, and 48% reported complete resolution.
The peripheral neuropathy is predominantly sensory; however, motor and sensorimotor peripheral neuropathy also occur. Monitor for symptoms of peripheral neuropathy such as hypoesthesia, hyperesthesia, paresthesia, dysesthesia, neuropathic pain, burning sensation, weakness, or gait disturbance. Patients experiencing new or worsening peripheral neuropathy may require a delay, dose reduction, or discontinuation of POLIVY [see Dosage and Administration (2.2)].
5.8 Embryo Fetal Toxicity (5.8 Embryo-Fetal Toxicity)
Based on the mechanism of action and findings from animal studies, POLIVY can cause fetal harm when administered to a pregnant woman. The small molecule component of POLIVY, MMAE, administered to rats caused adverse developmental outcomes, including embryo-fetal mortality and structural abnormalities, at exposures below those occurring clinically at the recommended dose.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with POLIVY and for 3 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with POLIVY and for 5 months after the last dose [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Peripheral Neuropathy: Monitor patients for peripheral neuropathy and modify or discontinue dose accordingly. (5.1)
- Infusion-Related Reactions: Premedicate with an antihistamine and antipyretic. Monitor patients closely during infusions. Interrupt or discontinue infusion for reactions. (5.2)
- Myelosuppression: Monitor complete blood counts. Manage using dose delays or reductions and growth factor support. Monitor for signs of infection. (5.3)
- Serious and Opportunistic Infections: Closely monitor patients for signs of bacterial, fungal, or viral infections. (5.4)
- Progressive Multifocal Leukoencephalopathy (PML): Monitor patients for new or worsening neurological, cognitive, or behavioral changes suggestive of PML. (5.5)
- Tumor Lysis Syndrome: Closely monitor patients with high tumor burden or rapidly proliferative tumors. (5.6)
- Hepatotoxicity: Monitor liver enzymes and bilirubin. (5.7)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for 3 months after the last dose. (5.8)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
- The recommended dose of POLIVY is 1.8 mg/kg as an intravenous infusion every 21 days for 6 cycles. (2)
- Administer the initial POLIVY dose over 90 minutes. Subsequent infusions may be administered over 30 minutes if the previous infusion is tolerated. (2)
- Premedicate with an antihistamine and antipyretic before POLIVY. (2)
- See Full Prescribing Information for instructions on preparation and administration. (2.4)
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
For injection: 30 mg/vial or 140 mg/vial of polatuzumab vedotin-piiq as a white to grayish-white lyophilized powder in a single-dose vial for reconstitution and further dilution.
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
5.2 Infusion Related Reactions (5.2 Infusion-Related Reactions)
POLIVY can cause infusion-related reactions, including severe cases. Delayed infusion-related reactions as late as 24 hours after receiving POLIVY have occurred.
With premedication, 13% of patients (58/435) in POLARIX reported infusion-related reactions after the administration of POLIVY plus R-CHP. The reactions were Grade 1 in 4.4% of patients, Grade 2 in 8%, and Grade 3 in 1.1%.
With premedication, 7% of patients (12/173) in Study GO29365 reported infusion-related reactions after the administration of POLIVY. The reactions were Grade 1 in 4.6% of patients, Grade 2 in 1.7%, and Grade 3 in 0.6%.
Symptoms occurring in ≥1% of patients included chills, dyspnea, pyrexia, pruritus, rash, and chest discomfort. Administer an antihistamine and antipyretic prior to the administration of POLIVY, and monitor patients closely throughout the infusion. If an infusion-related reaction occurs, interrupt the infusion and institute appropriate medical management [see Dosage and Administration (2.2)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described below reflect exposure to POLIVY 1.8 mg/kg in 480 patients with large B-cell lymphoma (LBCL), including those with previously untreated LBCL (POLARIX) and relapsed or refractory DLBCL (GO29365).
1.2 Relapsed Or Refractory Dlbcl, Nos (1.2 Relapsed or Refractory DLBCL, NOS)
POLIVY in combination with bendamustine and a rituximab product is indicated for the treatment of adult patients with relapsed or refractory DLBCL, NOS, after at least two prior therapies.
2.3 Recommended Prophylactic Medications
If not already premedicated for a rituximab product, administer an antihistamine and antipyretic at least 30 to 60 minutes prior to POLIVY for potential infusion-related reactions [see Warnings and Precautions (5.2)].
Administer prophylaxis for Pneumocystis jiroveci pneumonia and herpesvirus throughout treatment with POLIVY.
Administer prophylactic granulocyte colony-stimulating factor (G-CSF) for neutropenia in patients receiving POLIVY plus R-CHP. Consider prophylactic G-CSF administration for neutropenia in patients receiving POLIVY plus bendamustine and a rituximab product [see Warnings and Precautions (5.3)].
Administer tumor lysis syndrome prophylaxis for patients at increased risk of tumor lysis syndrome [see Warnings and Precautions (5.6)].
5.4 Serious and Opportunistic Infections
Fatal and/or serious infections, including opportunistic infections such as sepsis, pneumonia (including Pneumocystis jiroveci and other fungal pneumonia), herpesvirus infection, and cytomegalovirus infection have occurred in patients treated with POLIVY [see Adverse Reactions (6.1)].
In POLARIX, Grade 3–4 infections occurred in 14% (61/435) of patients treated with POLIVY plus R-CHP and infection-related deaths were reported in 1.1% of patients.
In Study GO29365, Grade 3 or higher infections occurred in 32% (55/173) of patients treated with POLIVYand infection-related deaths were reported in 2.9% of patients within 90 days of last treatment.
Closely monitor patients during treatment for signs of infection. Administer prophylaxis for Pneumocystis jiroveci pneumonia and herpesvirus. Administer prophylactic G-CSF for neutropenia as recommended [see Dosage and Administration (2.3)].
1.1 Previously Untreated Dlbcl, Nos Or Hgbl (1.1 Previously Untreated DLBCL, NOS or HGBL)
POLIVY in combination with a rituximab product, cyclophosphamide, doxorubicin, and prednisone (R-CHP) is indicated for the treatment of adult patients who have previously untreated diffuse large B-cell lymphoma (DLBCL), not otherwise specified (NOS) or high-grade B-cell lymphoma (HGBL) and who have an International Prognostic Index score of 2 or greater.
Principal Display Panel 30 Mg Vial Carton (PRINCIPAL DISPLAY PANEL - 30 mg Vial Carton)
NDC 50242-103-01
Polivy®
(polatuzumab vedotin-piiq)
For Injection
30 mg per vial
For Intravenous Infusion Only.
Reconstitute and Dilute
prior to administration.
Single-Dose Vial.
Discard unused portion.
CAUTION: Hazardous Agent
Rx only
1 vial
Genentech
11008718
Principal Display Panel 140 Mg Vial Carton (PRINCIPAL DISPLAY PANEL - 140 mg Vial Carton)
NDC 50242-105-01
Polivy®
(polatuzumab vedotin-piiq)
For Injection
140 mg per vial
For Intravenous Infusion Only.
Reconstitute and Dilute
prior to administration.
Single-Dose Vial.
Discard unused portion.
CAUTION: Hazardous Agent
Rx only
1 vial
Genentech
11007928
8.3 Females and Males of Reproductive Potential
POLIVY can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
2.4 Instructions for Preparation and Administration
Reconstitute and further dilute POLIVY prior to intravenous infusion.
POLIVY is a hazardous drug. Follow applicable special handling and disposal procedures.1
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
5.5 Progressive Multifocal Leukoencephalopathy (pml) (5.5 Progressive Multifocal Leukoencephalopathy (PML))
PML has been reported after treatment with POLIVY plus bendamustine and obinutuzumab in study GO29365 (0.6%, 1/173). Monitor for new or worsening neurological, cognitive, or behavioral changes. Hold POLIVY and any concomitant chemotherapy if PML is suspected, and permanently discontinue if the diagnosis is confirmed.
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies in animals have not been performed with polatuzumab vedotin-piiq or MMAE.
MMAE was positive for genotoxicity in the in vivo rat bone marrow micronucleus study through an aneugenic mechanism. MMAE was not mutagenic in the bacterial reverse mutation (Ames) assay or the L5178Y mouse lymphoma forward mutation assay.
Fertility studies in animals have not been performed with polatuzumab vedotin-piiq or MMAE. However, results of repeat-dose toxicity indicate the potential for polatuzumab vedotin-piiq to impair female and male fertility. In the 4-week repeat-dose toxicity study in rats with weekly dosing of 2, 6, and 10 mg/kg, dose-dependent testicular seminiferous tubule degeneration with abnormal lumen contents in the epididymis was observed. Findings in the testes and epididymis did not reverse and correlated with decreased testes weight and gross findings of small and/or soft testes at recovery necropsy in males given doses ≥2 mg/kg (below the exposure at the recommended dose based on unconjugated MMAE AUC).
MMAE-containing ADCs have been associated with adverse ovarian effects when administered to sexually immature animals. Adverse effects included decrease in, or absence of, secondary and tertiary ovarian follicles after weekly administration to cynomolgus monkeys in studies of 4-week duration. These effects showed a trend towards recovery 6 weeks after the end of dosing; no changes were observed in primordial follicles.
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:51:11.966299 · Updated: 2026-03-14T22:39:36.817935