Tofidence
1ab3207a-ebfe-4565-9ce5-3a1538815c83
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
TOFIDENCE ® (tocilizumab-bavi) is an interleukin-6 (IL-6) receptor antagonist indicated for treatment of: Rheumatoid Arthritis (RA) ( 1.1 ) Adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more Disease-Modifying Anti-Rheumatic Drugs (DMARDs). Giant Cell Arteritis (GCA) ( 1.2 ) Adult patients with giant cell arteritis. Polyarticular Juvenile Idiopathic Arthritis (PJIA) ( 1.3 ) Patients 2 years of age and older with active polyarticular juvenile idiopathic arthritis. Systemic Juvenile Idiopathic Arthritis (SJIA) ( 1.4 ) Patients 2 years of age and older with active systemic juvenile idiopathic arthritis. Coronavirus Disease 2019 (COVID-19) ( 1.5 ) Hospitalized adult patients with coronavirus disease 2019 (COVID-19) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
Dosage and Administration
For RA, pJIA and sJIA, TOFIDENCE may be used alone or in combination with methotrexate; and in RA, other non-biologic DMARDs may be used. ( 2 ) General Administration and Dosing Information ( 2.1 ) RA, GCA, PJIA and SJIA - It is recommended that TOFIDENCE not be initiated in patients with an absolute neutrophil count (ANC) below 2000 per mm 3 , platelet count below 100,000 per mm 3 , or ALT or AST above 1.5 times the upper limit of normal (ULN). ( 5.3 , 5.4 ) COVID-19 - It is recommended that TOFIDENCE not be initiated in patients with an absolute neutrophil count (ANC) below 1000 per mm 3 , platelet count below 50,000 mm3 , or ALT or AST above 10 times ULN. ( 5.3 , 5.4 ) In RA or COVID-19 patients, TOFIDENCE doses exceeding 800 mg per infusion are not recommended. ( 2.2 , 12.3 ) In GCA patients, TOFIDENCE doses exceeding 600 mg per infusion are not recommended. ( 2.3 , 12.3 ) Rheumatoid Arthritis ( 2.2 ) Recommended Adult Intravenous Dosage: When used in combination with non-biologic DMARDs or as monotherapy the recommended starting dose is 4 mg per kg every 4 weeks followed by an increase to 8 mg per kg every 4 weeks based on clinical response. Giant Cell Arteritis ( 2.3 ) Recommended Adult Intravenous Dosage: The recommended dose is 6 mg per kg every 4 weeks in combination with a tapering course of glucocorticoids. TOFIDENCE can be used alone following discontinuation of glucocorticoids. Polyarticular Juvenile Idiopathic Arthritis ( 2.4 ) Recommended Intravenous PJIA Dosage Every 4 Weeks Patients less than 30 kg weight 10 mg per kg Patients at or above 30 kg weight 8 mg per kg Systemic Juvenile Idiopathic Arthritis ( 2.5 ) Recommended Intravenous SJIA Dosage Every 2 Weeks Patients less than 30 kg weight 12 mg per kg Patients at or above 30 kg weight 8 mg per kg Coronavirus Disease 2019 ( 2.6 ) The recommended dosage of TOFIDENCE for adult patients with COVID-19 is 8 mg per kg administered by a 60-minute intravenous infusion. Administration of Intravenous Formulation ( 2.7 ) For patients with RA, GCA, COVID-19, PJIA, and SJIA patients at or above 30 kg, dilute to 100 mL in 0.9% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique. For PJIA and SJIA patients less than 30 kg, dilute to 50 mL in 0.9% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique. Administer as a single intravenous drip infusion over 1 hour; do not administer as bolus or push. Dose Modifications ( 2.8 ) Recommended for management of certain dose-related laboratory changes including elevated liver enzymes, neutropenia, and thrombocytopenia.
Contraindications
TOFIDENCE is contraindicated in patients with known hypersensitivity to tocilizumab products [see Warnings and Precautions (5.6) ].
Warnings and Precautions
Serious Infections – do not administer TOFIDENCE during an active infection, including localized infections. If a serious infection develops, interrupt TOFIDENCE until the infection is controlled. ( 5.1 ) Gastrointestinal (GI) perforation—use with caution in patients who may be at increased risk. ( 5.2 ) Hepatotoxicity- Monitor patients for signs and symptoms of hepatic injury. Modify or discontinue TOFIDENCE if abnormal liver tests persist or worsen or if clinical signs and symptoms of liver disease develop. ( 2.8 , 5.3 ) Laboratory monitoring—recommended due to potential consequences of treatment-related changes in neutrophils, platelets, lipids, and liver function tests. ( 2.8 , 5.4 ) Hypersensitivity reactions, including anaphylaxis and death and serious cutaneous reactions including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) – discontinue TOFIDENCE, treat promptly, and monitor until reaction resolves. ( 5.6 ) Live vaccines—Avoid use with TOFIDENCE. ( 5.9 , 7.3 )
Adverse Reactions
The following serious adverse reactions are described elsewhere in labeling: Serious Infections [see Warnings and Precautions (5.1) ] Gastrointestinal Perforations [see Warnings and Precautions (5.2) ] Laboratory Parameters [see Warnings and Precautions (5.4) ] Immunosuppression [see Warnings and Precautions (5.5) ] Hypersensitivity Reactions, Including Anaphylaxis [see Warnings and Precautions (5.6) ] Demyelinating Disorders [see Warnings and Precautions (5.7) ] Active Hepatic Disease and Hepatic Impairment [see Warnings and Precautions (5.8) ] Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
How Supplied
TOFIDENCE (tocilizumab-bavi) injection is a preservative-free, sterile, clear to opalescent, colorless to light yellow solution. TOFIDENCE is supplied as 80 mg/4 mL (NDC 78206-200-01), 200 mg/10 mL (NDC 78206-201-01), and 400 mg/20 mL (NDC 78206-202-01) individually packaged 20 mg/mL single-dose vials for further dilution prior to intravenous infusion.
Storage and Handling
TOFIDENCE (tocilizumab-bavi) injection is a preservative-free, sterile, clear to opalescent, colorless to light yellow solution. TOFIDENCE is supplied as 80 mg/4 mL (NDC 78206-200-01), 200 mg/10 mL (NDC 78206-201-01), and 400 mg/20 mL (NDC 78206-202-01) individually packaged 20 mg/mL single-dose vials for further dilution prior to intravenous infusion.
Description
Patients treated with tocilizumab products including TOFIDENCE are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1) , Adverse Reactions (6.1) ]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. If a serious infection develops, interrupt TOFIDENCE until the infection is controlled. Reported infections include: Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients, except those with COVID-19, should be tested for latent tuberculosis before TOFIDENCE use and during therapy. Treatment for latent infection should be initiated prior to TOFIDENCE use. Invasive fungal infections, including candidiasis, aspergillosis, and pneumocystis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease. Bacterial, viral and other infections due to opportunistic pathogens. The risks and benefits of treatment with TOFIDENCE should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection. Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with TOFIDENCE, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1) ].
Medication Information
Warnings and Precautions
Serious Infections – do not administer TOFIDENCE during an active infection, including localized infections. If a serious infection develops, interrupt TOFIDENCE until the infection is controlled. ( 5.1 ) Gastrointestinal (GI) perforation—use with caution in patients who may be at increased risk. ( 5.2 ) Hepatotoxicity- Monitor patients for signs and symptoms of hepatic injury. Modify or discontinue TOFIDENCE if abnormal liver tests persist or worsen or if clinical signs and symptoms of liver disease develop. ( 2.8 , 5.3 ) Laboratory monitoring—recommended due to potential consequences of treatment-related changes in neutrophils, platelets, lipids, and liver function tests. ( 2.8 , 5.4 ) Hypersensitivity reactions, including anaphylaxis and death and serious cutaneous reactions including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) – discontinue TOFIDENCE, treat promptly, and monitor until reaction resolves. ( 5.6 ) Live vaccines—Avoid use with TOFIDENCE. ( 5.9 , 7.3 )
Indications and Usage
TOFIDENCE ® (tocilizumab-bavi) is an interleukin-6 (IL-6) receptor antagonist indicated for treatment of: Rheumatoid Arthritis (RA) ( 1.1 ) Adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more Disease-Modifying Anti-Rheumatic Drugs (DMARDs). Giant Cell Arteritis (GCA) ( 1.2 ) Adult patients with giant cell arteritis. Polyarticular Juvenile Idiopathic Arthritis (PJIA) ( 1.3 ) Patients 2 years of age and older with active polyarticular juvenile idiopathic arthritis. Systemic Juvenile Idiopathic Arthritis (SJIA) ( 1.4 ) Patients 2 years of age and older with active systemic juvenile idiopathic arthritis. Coronavirus Disease 2019 (COVID-19) ( 1.5 ) Hospitalized adult patients with coronavirus disease 2019 (COVID-19) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
Dosage and Administration
For RA, pJIA and sJIA, TOFIDENCE may be used alone or in combination with methotrexate; and in RA, other non-biologic DMARDs may be used. ( 2 ) General Administration and Dosing Information ( 2.1 ) RA, GCA, PJIA and SJIA - It is recommended that TOFIDENCE not be initiated in patients with an absolute neutrophil count (ANC) below 2000 per mm 3 , platelet count below 100,000 per mm 3 , or ALT or AST above 1.5 times the upper limit of normal (ULN). ( 5.3 , 5.4 ) COVID-19 - It is recommended that TOFIDENCE not be initiated in patients with an absolute neutrophil count (ANC) below 1000 per mm 3 , platelet count below 50,000 mm3 , or ALT or AST above 10 times ULN. ( 5.3 , 5.4 ) In RA or COVID-19 patients, TOFIDENCE doses exceeding 800 mg per infusion are not recommended. ( 2.2 , 12.3 ) In GCA patients, TOFIDENCE doses exceeding 600 mg per infusion are not recommended. ( 2.3 , 12.3 ) Rheumatoid Arthritis ( 2.2 ) Recommended Adult Intravenous Dosage: When used in combination with non-biologic DMARDs or as monotherapy the recommended starting dose is 4 mg per kg every 4 weeks followed by an increase to 8 mg per kg every 4 weeks based on clinical response. Giant Cell Arteritis ( 2.3 ) Recommended Adult Intravenous Dosage: The recommended dose is 6 mg per kg every 4 weeks in combination with a tapering course of glucocorticoids. TOFIDENCE can be used alone following discontinuation of glucocorticoids. Polyarticular Juvenile Idiopathic Arthritis ( 2.4 ) Recommended Intravenous PJIA Dosage Every 4 Weeks Patients less than 30 kg weight 10 mg per kg Patients at or above 30 kg weight 8 mg per kg Systemic Juvenile Idiopathic Arthritis ( 2.5 ) Recommended Intravenous SJIA Dosage Every 2 Weeks Patients less than 30 kg weight 12 mg per kg Patients at or above 30 kg weight 8 mg per kg Coronavirus Disease 2019 ( 2.6 ) The recommended dosage of TOFIDENCE for adult patients with COVID-19 is 8 mg per kg administered by a 60-minute intravenous infusion. Administration of Intravenous Formulation ( 2.7 ) For patients with RA, GCA, COVID-19, PJIA, and SJIA patients at or above 30 kg, dilute to 100 mL in 0.9% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique. For PJIA and SJIA patients less than 30 kg, dilute to 50 mL in 0.9% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique. Administer as a single intravenous drip infusion over 1 hour; do not administer as bolus or push. Dose Modifications ( 2.8 ) Recommended for management of certain dose-related laboratory changes including elevated liver enzymes, neutropenia, and thrombocytopenia.
Contraindications
TOFIDENCE is contraindicated in patients with known hypersensitivity to tocilizumab products [see Warnings and Precautions (5.6) ].
Adverse Reactions
The following serious adverse reactions are described elsewhere in labeling: Serious Infections [see Warnings and Precautions (5.1) ] Gastrointestinal Perforations [see Warnings and Precautions (5.2) ] Laboratory Parameters [see Warnings and Precautions (5.4) ] Immunosuppression [see Warnings and Precautions (5.5) ] Hypersensitivity Reactions, Including Anaphylaxis [see Warnings and Precautions (5.6) ] Demyelinating Disorders [see Warnings and Precautions (5.7) ] Active Hepatic Disease and Hepatic Impairment [see Warnings and Precautions (5.8) ] Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
Storage and Handling
TOFIDENCE (tocilizumab-bavi) injection is a preservative-free, sterile, clear to opalescent, colorless to light yellow solution. TOFIDENCE is supplied as 80 mg/4 mL (NDC 78206-200-01), 200 mg/10 mL (NDC 78206-201-01), and 400 mg/20 mL (NDC 78206-202-01) individually packaged 20 mg/mL single-dose vials for further dilution prior to intravenous infusion.
How Supplied
TOFIDENCE (tocilizumab-bavi) injection is a preservative-free, sterile, clear to opalescent, colorless to light yellow solution. TOFIDENCE is supplied as 80 mg/4 mL (NDC 78206-200-01), 200 mg/10 mL (NDC 78206-201-01), and 400 mg/20 mL (NDC 78206-202-01) individually packaged 20 mg/mL single-dose vials for further dilution prior to intravenous infusion.
Description
Patients treated with tocilizumab products including TOFIDENCE are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1) , Adverse Reactions (6.1) ]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids. If a serious infection develops, interrupt TOFIDENCE until the infection is controlled. Reported infections include: Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients, except those with COVID-19, should be tested for latent tuberculosis before TOFIDENCE use and during therapy. Treatment for latent infection should be initiated prior to TOFIDENCE use. Invasive fungal infections, including candidiasis, aspergillosis, and pneumocystis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease. Bacterial, viral and other infections due to opportunistic pathogens. The risks and benefits of treatment with TOFIDENCE should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection. Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with TOFIDENCE, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1) ].
Section 42229-5
Not Recommended for Concomitant Use with Biological DMARDs
Tocilizumab products have not been studied in combination with biological DMARDs such as TNF antagonists, IL-1R antagonists, anti-CD20 monoclonal antibodies and selective co-stimulation modulators because of the possibility of increased immunosuppression and increased risk of infection. Avoid using TOFIDENCE with biological DMARDs.
Section 42231-1
| Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: 07/2025 | |||
|
Medication Guide
TOFIDENCE® (TOE-FIH-DENCE) (tocilizumab-bavi) injection for intravenous use |
||||
|
What is the most important information I should know about TOFIDENCE?
Your healthcare provider should monitor you closely for signs and symptoms of TB during and after treatment with TOFIDENCE.
|
||||
|
|
|
||
|
||||
|
|
|||
You should not receive TOFIDENCE if your neutrophil or platelet counts are too low or your liver function tests are too high. Your healthcare provider may stop your TOFIDENCE treatment for a period of time or change your dose of medicine if needed because of changes in these blood test results.
|
||||
|
What is TOFIDENCE?
TOFIDENCE is a prescription medicine called an Interleukin-6 (IL-6) receptor antagonist. TOFIDENCE is used:
|
||||
| Do not take TOFIDENCE: if you are allergic to tocilizumab products, or any of the ingredients in TOFIDENCE. See the end of this Medication Guide for a complete list of ingredients in TOFIDENCE. | ||||
Before you receive TOFIDENCE, tell your healthcare provider about all of your medical conditions, including if you:
|
||||
|
Tell your healthcare provider about all of the medicines you take, including prescription, over-the-counter medicines, vitamins and herbal supplements. TOFIDENCE and other medicines may affect each other causing side effects. Especially tell your healthcare provider if you take:
|
||||
|
How will I receive TOFIDENCE?
Into a vein (IV or intravenous infusion) for Rheumatoid Arthritis, Giant Cell Arteritis, PJIA, SJIA, or COVID-19:
|
||||
|
What are the possible side effects with TOFIDENCE?
|
||||
|
|
|
||
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Organon USA LLC, a subsidiary of Organon & Co., at 1-844-674-3200. |
||||
|
General information about the safe and effective use of TOFIDENCE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not give TOFIDENCE to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TOFIDENCE that is written for health professionals. |
||||
|
What are the ingredients in TOFIDENCE?
Active ingredient: tocilizumab-bavi. Inactive ingredients of Intravenous TOFIDENCE: arginine hydrochloride, histidine, L-histidine hydrochloride monohydrate,polysorbate 80, sucrose, and water for Injection. Manufactured by: Organon USA LLC, a subsidiary of ORGANON & Co., Jersey City, NJ 07302, USA U.S. License No. 2331 © 2025 Organon group of companies. All rights reserved. For more information, go to www.tofidence.com or call 1-844-674-3200 . usmg-og9401-i-2507r000 |
||||
Section 43683-2
Section 44425-7
Storage and Handling: Do not use beyond expiration date on the container or package. TOFIDENCE must be refrigerated at 36°F to 46°F (2°C to 8°C). Do not freeze. Protect the vials from light by storage in the original package until time of use.
10 Overdosage
There are limited data available on overdoses with tocilizumab products. One case of accidental overdose was reported with intravenous tocilizumab in which a patient with multiple myeloma received a dose of 40 mg per kg. No adverse drug reactions were observed. No serious adverse drug reactions were observed in healthy volunteers who received single doses of up to 28 mg per kg, although all 5 patients at the highest dose of 28 mg per kg developed dose-limiting neutropenia.
In case of an overdose, it is recommended that the patient be monitored for signs and symptoms of adverse reactions. Patients who develop adverse reactions should receive appropriate symptomatic treatment.
11 Description
Tocilizumab-bavi is a recombinant humanized anti-human interleukin 6 (IL-6) receptor monoclonal antibody of the immunoglobulin IgG1κ (gamma 1, kappa) subclass with a typical H2L2 polypeptide structure. Each light chain and heavy chain consists of 214 and 448 amino acids, respectively. The four polypeptide chains are linked intra- and inter-molecularly by disulfide bonds. Tocilizumab-bavi has a molecular weight of approximately 148 kDa. The antibody is produced in mammalian (Chinese hamster ovary) cells.
5.9 Vaccinations
Avoid use of live vaccines concurrently with TOFIDENCE as clinical safety has not been established. No data are available on the secondary transmission of infection from persons receiving live vaccines to patients receiving tocilizumab products.
No data are available on the effectiveness of vaccination in patients receiving tocilizumab products. Because IL-6 inhibition may interfere with the normal immune response to new antigens, it is recommended that all patients, particularly pediatric or elderly patients, if possible, be brought up to date with all immunizations in agreement with current immunization guidelines prior to initiating TOFIDENCE therapy. The interval between live vaccinations and initiation of TOFIDENCE therapy should be in accordance with current vaccination guidelines regarding immunosuppressive agents.
7.3 Live Vaccines
Avoid use of live vaccines concurrently with TOFIDENCE [see Warnings and Precautions (5.9)].
8.4 Pediatric Use
TOFIDENCE by intravenous use is indicated for the treatment of pediatric patients with:
- Active systemic juvenile idiopathic arthritis in patients 2 years of age and older
- Active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older
The safety and effectiveness of TOFIDENCE in pediatric patients with conditions other than PJIA or SJIA have not been established. The safety and effectiveness in pediatric patients below the age of 2 have not been established in PJIA or SJIA.
8.5 Geriatric Use
Of the 2644 patients who received tocilizumab in Studies I to V [see Clinical Studies (14)], a total of 435 rheumatoid arthritis patients were 65 years of age and older, including 50 patients 75 years and older. The frequency of serious infection among tocilizumab treated subjects 65 years of age and older was higher than those under the age of 65. As there is a higher incidence of infections in the elderly population in general, caution should be used when treating the elderly.
In the EMPACTA, COVACTA, and REMDACTA studies, of the 974 COVID-19 patients in the tocilizumab arm, 375 (39%) were 65 years of age or older. No overall differences in safety or effectiveness of tocilizumab were observed between patients 65 years of age and older and those under the age of 65 years of age in these studies [see Adverse Reactions (6.5) and Clinical Studies (14.5)].
In the RECOVERY study, of the 2022 COVID-19 patients in the tocilizumab arm, 930 (46%) were 65 years of age or older. No overall differences in effectiveness of tocilizumab were observed between patients 65 years of age and older and those under the age 65 years of age in this study [see Clinical Studies (14.5)].
5.3 Hepatotoxicity
Serious cases of hepatic injury have been observed in patients taking intravenous tocilizumab products. Some of these cases have resulted in liver transplant or death. Time to onset for cases ranged from months to years after treatment initiation with tocilizumab products. While most cases presented with marked elevations of transaminases (> 5 times ULN), some cases presented with signs or symptoms of liver dysfunction and only mildly elevated transaminases.
During randomized controlled studies, treatment with tocilizumab was associated with a higher incidence of transaminase elevations [see Adverse Reactions (6.1, 6.3, 6.4)]. Increased frequency and magnitude of these elevations was observed when potentially hepatotoxic drugs (e.g., MTX) were used in combination with tocilizumab.
For RA and GCA patients, obtain a liver test panel (serum alanine aminotransferase [ALT], aspartate aminotransferase [AST], alkaline phosphatase, and total bilirubin) before initiating TOFIDENCE, every 4 to 8 weeks after start of therapy for the first 6 months of treatment and every 3 months thereafter. It is not recommended to initiate TOFIDENCE treatment in RA or GCA patients with elevated transaminases ALT or AST greater than 1.5x ULN. In patients who develop elevated ALT or AST greater than 5x ULN, discontinue TOFIDENCE. For recommended modifications based upon increase in transaminases [see Dosage and Administration (2.8)].
Patients hospitalized with COVID-19 may have elevated ALT or AST levels. Multi-organ failure with involvement of the liver is recognized as a complication of severe COVID-19. The decision to administer TOFIDENCE should balance the potential benefit of treating COVID-19 against the potential risks of acute treatment with TOFIDENCE. It is not recommended to initiate TOFIDENCE treatment in COVID-19 patients with elevated ALT or AST above 10 x ULN. Monitor ALT and AST during treatment.
Measure liver tests promptly in patients who report symptoms that may indicate liver injury, such as fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice. In this clinical context, if the patient is found to have abnormal liver tests (e.g., ALT greater than three times the upper limit of the reference range, serum total bilirubin greater than two times the upper limit of the reference range), TOFIDENCE treatment should be interrupted and investigation done to establish the probable cause. TOFIDENCE should only be restarted in patients with another explanation for the liver test abnormalities after normalization of the liver tests.
A similar pattern of liver enzyme elevation is noted with tocilizumab products treatment in the PJIA and SJIA populations. Monitor liver test panel at the time of the second administration and thereafter every 4 to 8 weeks for PJIA and every 2 to 4 weeks for SJIA.
4 Contraindications
TOFIDENCE is contraindicated in patients with known hypersensitivity to tocilizumab products [see Warnings and Precautions (5.6)].
6 Adverse Reactions
The following serious adverse reactions are described elsewhere in labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Gastrointestinal Perforations [see Warnings and Precautions (5.2)]
- Laboratory Parameters [see Warnings and Precautions (5.4)]
- Immunosuppression [see Warnings and Precautions (5.5)]
- Hypersensitivity Reactions, Including Anaphylaxis [see Warnings and Precautions (5.6)]
- Demyelinating Disorders [see Warnings and Precautions (5.7)]
- Active Hepatic Disease and Hepatic Impairment [see Warnings and Precautions (5.8)]
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
8.7 Renal Impairment
No dose adjustment is required in patients with mild or moderate renal impairment. Tocilizumab products have not been studied in patients with severe renal impairment [see Clinical Pharmacology (12.3)].
12.2 Pharmacodynamics
In clinical studies in RA patients with the 4 mg per kg and 8 mg per kg intravenous doses decreases in levels of C-reactive protein (CRP) to within normal ranges were seen as early as week 2. Changes in pharmacodynamic parameters were observed (i.e., decreases in rheumatoid factor, erythrocyte sedimentation rate (ESR), serum amyloid A, fibrinogen and increases in hemoglobin) with doses, however the greatest improvements were observed with 8 mg per kg tocilizumab. Pharmacodynamic changes were also observed to occur after tocilizumab administration in GCA, PJIA, and SJIA patients (decreases in CRP, ESR, and increases in hemoglobin). The relationship between these pharmacodynamic findings and clinical efficacy is not known.
In healthy subjects administered tocilizumab in doses from 2 to 28 mg per kg intravenously, absolute neutrophil counts decreased to the nadir 3 to 5 days following tocilizumab administration. Thereafter, neutrophils recovered towards baseline in a dose dependent manner. Rheumatoid arthritis and GCA patients demonstrated a similar pattern of absolute neutrophil counts following tocilizumab administration [see Warnings and Precautions (5.4)].
12.3 Pharmacokinetics
PK of tocilizumab is characterized by nonlinear elimination which is a combination of linear clearance and Michaelis-Menten elimination. The nonlinear part of tocilizumab elimination leads to an increase in exposure that is more than dose-proportional. The pharmacokinetic parameters of tocilizumab do not change with time. Due to the dependence of total clearance on tocilizumab serum concentrations, the half-life of tocilizumab is also concentration-dependent and varies depending on the serum concentration level. Population pharmacokinetic analyses in any patient population tested so far indicate no relationship between apparent clearance and the presence of anti-drug antibodies.
5.5 Immunosuppression
The impact of treatment with tocilizumab products on the development of malignancies is not known but malignancies were observed in clinical studies [see Adverse Reactions (6.1)]. TOFIDENCE is an immunosuppressant, and treatment with immunosuppressants may result in an increased risk of malignancies.
5.1 Serious Infections
Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, protozoal, or other opportunistic pathogens have been reported in patients receiving immunosuppressive agents including tocilizumab products. The most common serious infections included pneumonia, urinary tract infection, cellulitis, herpes zoster, gastroenteritis, diverticulitis, sepsis and bacterial arthritis [see Adverse Reactions (6.1)]. Among opportunistic infections, tuberculosis, cryptococcus, aspergillosis, candidiasis, and pneumocystosis were reported with tocilizumab products. Other serious infections, not reported in clinical studies, may also occur (e.g., histoplasmosis, coccidioidomycosis, listeriosis). Patients have presented with disseminated rather than localized disease, and were often taking concomitant immunosuppressants such as methotrexate or corticosteroids which in addition to rheumatoid arthritis may predispose them to infections.
Do not administer TOFIDENCE in patients with an active infection, including localized infections. The risks and benefits of treatment should be considered prior to initiating TOFIDENCE in patients:
- with chronic or recurrent infection;
- who have been exposed to tuberculosis;
- with a history of serious or an opportunistic infection;
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
- with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with TOFIDENCE, as signs and symptoms of acute inflammation may be lessened due to suppression of the acute phase reactants [see Dosage and Administration (2.1), Adverse Reactions (6.1), and Patient Counseling Information (17)].
Hold TOFIDENCE if a patient develops a serious infection, an opportunistic infection, or sepsis. A patient who develops a new infection during treatment with TOFIDENCE should undergo a prompt and complete diagnostic workup appropriate for an immunocompromised patient, initiate appropriate antimicrobial therapy, and closely monitor the patient.
8.6 Hepatic Impairment
The safety and efficacy of tocilizumab products have not been studied in patients with hepatic impairment, including patients with positive HBV and HCV serology [see Warnings and Precautions (5.8)].
1 Indications and Usage
TOFIDENCE® (tocilizumab-bavi) is an interleukin-6 (IL-6) receptor antagonist indicated for treatment of:
Rheumatoid Arthritis (RA) (1.1)
- Adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more Disease-Modifying Anti-Rheumatic Drugs (DMARDs).
Giant Cell Arteritis (GCA) (1.2)
- Adult patients with giant cell arteritis.
Polyarticular Juvenile Idiopathic Arthritis (PJIA) (1.3)
- Patients 2 years of age and older with active polyarticular juvenile idiopathic arthritis.
Systemic Juvenile Idiopathic Arthritis (SJIA) (1.4)
- Patients 2 years of age and older with active systemic juvenile idiopathic arthritis.
Coronavirus Disease 2019 (COVID-19) (1.5)
- Hospitalized adult patients with coronavirus disease 2019 (COVID-19) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
12.1 Mechanism of Action
Tocilizumab products bind to both soluble and membrane-bound IL-6 receptors (sIL-6R and mIL-6R), and have been shown to inhibit IL-6-mediated signaling through these receptors. IL-6 is a pleiotropic pro-inflammatory cytokine produced by a variety of cell types including T- and B-cells, lymphocytes, monocytes and fibroblasts. IL-6 has been shown to be involved in diverse physiological processes such as T-cell activation, induction of immunoglobulin secretion, initiation of hepatic acute phase protein synthesis, and stimulation of hematopoietic precursor cell proliferation and differentiation. IL-6 is also produced by synovial and endothelial cells leading to local production of IL-6 in joints affected by inflammatory processes such as rheumatoid arthritis.
5 Warnings and Precautions
- Serious Infections – do not administer TOFIDENCE during an active infection, including localized infections. If a serious infection develops, interrupt TOFIDENCE until the infection is controlled. (5.1)
- Gastrointestinal (GI) perforation—use with caution in patients who may be at increased risk. (5.2)
- Hepatotoxicity- Monitor patients for signs and symptoms of hepatic injury. Modify or discontinue TOFIDENCE if abnormal liver tests persist or worsen or if clinical signs and symptoms of liver disease develop. (2.8, 5.3)
- Laboratory monitoring—recommended due to potential consequences of treatment-related changes in neutrophils, platelets, lipids, and liver function tests. (2.8, 5.4)
- Hypersensitivity reactions, including anaphylaxis and death and serious cutaneous reactions including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) – discontinue TOFIDENCE, treat promptly, and monitor until reaction resolves. (5.6)
- Live vaccines—Avoid use with TOFIDENCE. (5.9, 7.3)
2 Dosage and Administration
For RA, pJIA and sJIA, TOFIDENCE may be used alone or in combination with methotrexate; and in RA, other non-biologic DMARDs may be used. (2)
General Administration and Dosing Information (2.1)
- RA, GCA, PJIA and SJIA - It is recommended that TOFIDENCE not be initiated in patients with an absolute neutrophil count (ANC) below 2000 per mm3, platelet count below 100,000 per mm3, or ALT or AST above 1.5 times the upper limit of normal (ULN). (5.3, 5.4)
- COVID-19 - It is recommended that TOFIDENCE not be initiated in patients with an absolute neutrophil count (ANC) below 1000 per mm3 , platelet count below 50,000 mm3 , or ALT or AST above 10 times ULN. (5.3, 5.4)
- In RA or COVID-19 patients, TOFIDENCE doses exceeding 800 mg per infusion are not recommended. (2.2, 12.3)
- In GCA patients, TOFIDENCE doses exceeding 600 mg per infusion are not recommended. (2.3, 12.3)
Rheumatoid Arthritis (2.2)
Recommended Adult Intravenous Dosage:
When used in combination with non-biologic DMARDs or as monotherapy the recommended starting dose is 4 mg per kg every 4 weeks followed by an increase to 8 mg per kg every 4 weeks based on clinical response.
Giant Cell Arteritis (2.3)
Recommended Adult Intravenous Dosage:
The recommended dose is 6 mg per kg every 4 weeks in combination with a tapering course of glucocorticoids. TOFIDENCE can be used alone following discontinuation of glucocorticoids.
Polyarticular Juvenile Idiopathic Arthritis (2.4)
| Recommended Intravenous PJIA Dosage Every 4 Weeks | |
|---|---|
| Patients less than 30 kg weight | 10 mg per kg |
| Patients at or above 30 kg weight | 8 mg per kg |
Systemic Juvenile Idiopathic Arthritis (2.5)
| Recommended Intravenous SJIA Dosage Every 2 Weeks | |
|---|---|
| Patients less than 30 kg weight | 12 mg per kg |
| Patients at or above 30 kg weight | 8 mg per kg |
Coronavirus Disease 2019 (2.6)
The recommended dosage of TOFIDENCE for adult patients with COVID-19 is 8 mg per kg administered by a 60-minute intravenous infusion.
Administration of Intravenous Formulation (2.7)
- For patients with RA, GCA, COVID-19, PJIA, and SJIA patients at or above 30 kg, dilute to 100 mL in 0.9% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique.
- For PJIA and SJIA patients less than 30 kg, dilute to 50 mL in 0.9% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique.
- Administer as a single intravenous drip infusion over 1 hour; do not administer as bolus or push.
Dose Modifications (2.8)
- Recommended for management of certain dose-related laboratory changes including elevated liver enzymes, neutropenia, and thrombocytopenia.
5.7 Demyelinating Disorders
The impact of treatment with tocilizumab products on demyelinating disorders is not known, but multiple sclerosis and chronic inflammatory demyelinating polyneuropathy were reported rarely in RA clinical studies. Monitor patients for signs and symptoms potentially indicative of demyelinating disorders. Prescribers should exercise caution in considering the use of TOFIDENCE in patients with preexisting or recent onset demyelinating disorders.
9 Drug Abuse and Dependence
No studies on the potential for tocilizumab products to cause dependence have been performed. However, there is no evidence from the available data that tocilizumab products treatment results in dependence.
3 Dosage Forms and Strengths
Intravenous Infusion
Injection: 80 mg/4 mL (20 mg/mL), 200 mg/10 mL (20 mg/mL), 400 mg/20 mL (20 mg/mL) in single-dose vials for further dilution prior to intravenous infusion (3)
6.6 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of tocilizumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hypersensitivity Reactions: Fatal anaphylaxis, Stevens-Johnson Syndrome, Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) [see Warnings and Precautions (5.6)]
- Pancreatitis
- Drug-induced liver injury, Hepatitis, Hepatic failure, Jaundice [see Warnings and Precautions (5.3)]
1.1 Rheumatoid Arthritis (ra)
TOFIDENCE (tocilizumab-bavi) is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more Disease-Modifying Anti-Rheumatic Drugs (DMARDs).
8 Use in Specific Populations
1.2 Giant Cell Arteritis (gca)
TOFIDENCE® (tocilizumab-bavi) is indicated for the treatment of giant cell arteritis (GCA) in adult patients.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
5.2 Gastrointestinal Perforations
Events of gastrointestinal perforation have been reported in clinical trials, primarily as complications of diverticulitis in patients treated with tocilizumab. Use TOFIDENCE with caution in patients who may be at increased risk for gastrointestinal perforation. Promptly evaluate patients presenting with fever, new onset abdominal symptoms, and a change in bowel habits for early identification of gastrointestinal perforation [see Adverse Reactions (6.1)].
Warning: Risk of Serious Infections
Patients treated with tocilizumab products including TOFIDENCE are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt TOFIDENCE until the infection is controlled.
Reported infections include:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients, except those with COVID-19, should be tested for latent tuberculosis before TOFIDENCE use and during therapy. Treatment for latent infection should be initiated prior to TOFIDENCE use.
- Invasive fungal infections, including candidiasis, aspergillosis, and pneumocystis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral and other infections due to opportunistic pathogens.
The risks and benefits of treatment with TOFIDENCE should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with TOFIDENCE, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1)].
16 How Supplied/storage and Handling
TOFIDENCE (tocilizumab-bavi) injection is a preservative-free, sterile, clear to opalescent, colorless to light yellow solution. TOFIDENCE is supplied as 80 mg/4 mL (NDC 78206-200-01), 200 mg/10 mL (NDC 78206-201-01), and 400 mg/20 mL (NDC 78206-202-01) individually packaged 20 mg/mL single-dose vials for further dilution prior to intravenous infusion.
1.5 Coronavirus Disease 2019 (covid 19)
TOFIDENCE® (tocilizumab-bavi) is indicated for the treatment of coronavirus disease 2019 (COVID-19) in hospitalized adult patients who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
2.6 Coronavirus Disease 2019 (covid 19)
Administer TOFIDENCE by intravenous infusion only.
The recommended dosage of TOFIDENCE for treatment of adult patients with COVID-19 is 8 mg per kg administered as a single 60-minute intravenous infusion. If clinical signs or symptoms worsen or do not improve after the first dose, one additional infusion of TOFIDENCE may be administered at least 8 hours after the initial infusion.
- Doses exceeding 800 mg per infusion are not recommended in patients with COVID-19.
7.2 Interactions With Cyp450 Substrates
Cytochrome P450s in the liver are down-regulated by infection and inflammation stimuli including cytokines such as IL-6. Inhibition of IL-6 signaling in RA patients treated with tocilizumab products may restore CYP450 activities to higher levels than those in the absence of tocilizumab products leading to increased metabolism of drugs that are CYP450 substrates. In vitro studies showed that tocilizumab has the potential to affect expression of multiple CYP enzymes including CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. Its effect on CYP2C8 or transporters is unknown. In vivo studies with omeprazole, metabolized by CYP2C19 and CYP3A4, and simvastatin, metabolized by CYP3A4, showed up to a 28% and 57% decrease in exposure one week following a single dose of tocilizumab, respectively. The effect of tocilizumab products on CYP enzymes may be clinically relevant for CYP450 substrates with narrow therapeutic index, where the dose is individually adjusted. Upon initiation or discontinuation of TOFIDENCE, in patients being treated with these types of medicinal products, perform therapeutic monitoring of effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) and the individual dose of the medicinal product adjusted as needed. Exercise caution when coadministering TOFIDENCE with CYP3A4 substrate drugs where decrease in effectiveness is undesirable, e.g., oral contraceptives, lovastatin, atorvastatin, etc. The effect of tocilizumab products on CYP450 enzyme activity may persist for several weeks after stopping therapy [see Clinical Pharmacology (12.3)].
14.5 Covid 19 Intravenous Administration
The efficacy of tocilizumab for the treatment of COVID-19 was based on RECOVERY (NCT04381936), a randomized, controlled, open-label, platform study, and supported by the results from EMPACTA (NCT04372186), a randomized, double-blind, placebo-controlled study. Results of two other randomized, double-blind, placebo-controlled studies, COVACTA (NCT04320615) and REMDACTA (NCT04409262), which evaluated the efficacy of tocilizumab for the treatment of COVID-19 are also summarized.
2.2 Recommended Dosage for Rheumatoid Arthritis
TOFIDENCE may be used as monotherapy or concomitantly with methotrexate or other non-biologic DMARDs as an intravenous infusion.
Principal Display Panel 80 Mg/4 Ml Vial Carton
Rx Only
NDC 78206-200-01
Tofidence®
(tocilizumab-bavi)
Injection
80 mg/4 mL
(20 mg/mL)
For Intravenous Infusion
after dilution
Single-Dose Vial
Discard unused portion.
ORGANON
1.4 Systemic Juvenile Idiopathic Arthritis (sjia)
TOFIDENCE® (tocilizumab-bavi) is indicated for the treatment of active systemic juvenile idiopathic arthritis in patients 2 years of age and older.
5.8 Active Hepatic Disease and Hepatic Impairment
Treatment with TOFIDENCE is not recommended in patients with active hepatic disease or hepatic impairment [see Adverse Reactions (6.1), Use in Specific Populations (8.6)].
Principal Display Panel 200 Mg/10 Ml Vial Carton
Rx Only
NDC 78206-201-01
Tofidence®
(tocilizumab-bavi)
Injection
200 mg/10 mL
(20 mg/mL)
For Intravenous Infusion
after dilution
Single-Dose Vial
Discard unused portion.
ORGANON
Principal Display Panel 400 Mg/20 Ml Vial Carton
Rx Only
NDC 78206-202-01
Tofidence®
(tocilizumab-bavi)
Injection
400 mg/20 mL
(20 mg/mL)
For Intravenous Infusion
after dilution
Single-Dose Vial
Discard unused portion.
ORGANON
5.6 Hypersensitivity Reactions, Including Anaphylaxis
Hypersensitivity reactions, including anaphylaxis, have been reported in association with tocilizumab products and anaphylactic events with a fatal outcome have been reported with intravenous infusion of tocilizumab products. Anaphylaxis and other hypersensitivity reactions that required treatment discontinuation were reported in 0.1% (3 out of 2644) of patients in the 6-month controlled trials of intravenous tocilizumab and 0.2% (8 out of 4009) of patients in the intravenous all-exposure RA population. In the SJIA controlled trial with intravenous tocilizumab, 1 out of 112 patients (0.9%) experienced hypersensitivity reactions that required treatment discontinuation. In the PJIA controlled trial with intravenous tocilizumab 0 out of 188 patients (0%) in the tocilizumab all-exposure population experienced hypersensitivity reactions that required treatment discontinuation. Reactions that required treatment discontinuation included generalized erythema, rash, and urticaria.
In the postmarketing setting, events of hypersensitivity reactions, including anaphylaxis and death have occurred in patients treated with a range of doses of intravenous tocilizumab products, with or without concomitant therapies. Events have occurred in patients who received premedication. Hypersensitivity, including anaphylaxis events, have occurred both with and without previous hypersensitivity reactions and as early as the first infusion of tocilizumab products [see Adverse Reactions (6.6)]. In addition, serious cutaneous reactions, including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), have been reported in patients with autoinflammatory conditions treated with tocilizumab products.
TOFIDENCE for intravenous use should only be infused by a healthcare professional with appropriate medical support to manage anaphylaxis. If a hypersensitivity reaction occurs, immediately discontinue TOFIDENCE, treat promptly and monitor until signs and symptoms resolve.
1.3 Polyarticular Juvenile Idiopathic Arthritis (pjia)
TOFIDENCE® (tocilizumab-bavi) is indicated for the treatment of active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older.
14.1 Rheumatoid Arthritis—intravenous Administration
The efficacy and safety of intravenously administered tocilizumab was assessed in five randomized, double-blind, multicenter studies in patients greater than 18 years with active rheumatoid arthritis diagnosed according to American College of Rheumatology (ACR) criteria. Patients had at least 8 tender and 6 swollen joints at baseline. Tocilizumab was given intravenously every 4 weeks as monotherapy (Study I), in combination with methotrexate (MTX) (Studies II and III) or other disease-modifying anti-rheumatic drugs (DMARDs) (Study IV) in patients with an inadequate response to those drugs, or in combination with MTX in patients with an inadequate response to TNF antagonists (Study V).
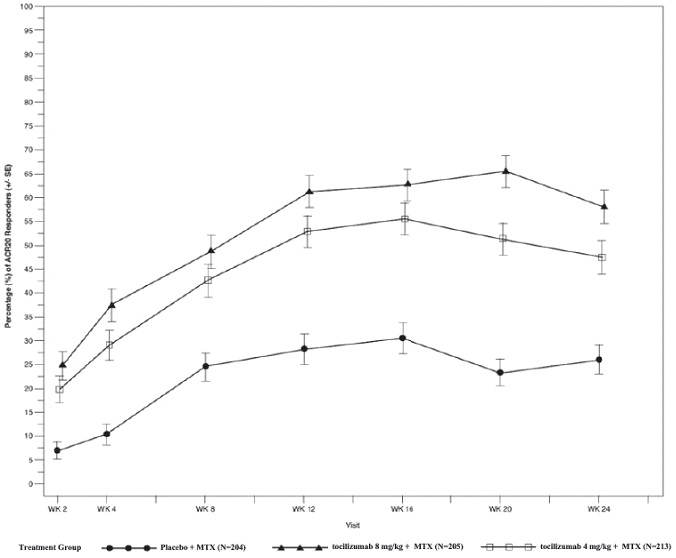
Study I (NCT00109408) evaluated patients with moderate to severe active rheumatoid arthritis who had not been treated with MTX within 24 weeks prior to randomization, or who had not discontinued previous methotrexate treatment as a result of clinically important toxic effects or lack of response. In this study, 67% of patients were MTX-naïve, and over 40% of patients had rheumatoid arthritis less than 2 years. Patients received tocilizumab 8 mg per kg monotherapy or MTX alone (dose titrated over 8 weeks from 7.5 mg to a maximum of 20 mg weekly). The primary endpoint was the proportion of tocilizumab patients who achieved an ACR 20 response at Week 24.
Study II (NCT00106535) was a 104-week study with an optional 156-week extension phase that evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response to MTX. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). Upon completion of 52-weeks, patients received open-label treatment with tocilizumab 8 mg per kg through 104 weeks or they had the option to continue their double-blind treatment if they maintained a greater than 70% improvement in swollen/tender joint count. Two pre-specified interim analyses at week 24 and week 52 were conducted. The primary endpoint at week 24 was the proportion of patients who achieved an ACR 20 response. At weeks 52 and 104, the primary endpoints were change from baseline in modified total Sharp-Genant score and the area under the curve (AUC) of the change from baseline in HAQ-DI score.
Study III (NCT00106548) evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response to MTX. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
Study IV (NCT00106574) evaluated patients who had an inadequate response to their existing therapy, including one or more DMARDs. Patients received tocilizumab 8 mg per kg or placebo every four weeks, in combination with the stable DMARDs. The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
Study V (NCT00106522) evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response or were intolerant to one or more TNF antagonist therapies. The TNF antagonist therapy was discontinued prior to randomization. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
14.2 Giant Cell Arteritis Intravenous Administration
Intravenously administered tocilizumab in patients with GCA was assessed in WP41152 (NCT03923738), an open-label PK-PD and safety study to determine the appropriate intravenous dose of tocilizumab that achieved comparable PK-PD profiles to tocilizumab by another route of administration.
At enrollment, all patients (n=24) were in remission on tocilizumab IV. In Period 1, all patients received open-label tocilizumab IV 7 mg/kg every 4 weeks for 20 weeks. Patients who completed Period 1 and remained in remission (n=22) were eligible to enter Period 2, and received open-label tocilizumab IV 6 mg/kg every 4 weeks for 20 weeks.
The efficacy of intravenous tocilizumab 6 mg/kg in adult patients with GCA is based on pharmacokinetic exposure and extrapolation to the efficacy established for tocilizumab by another route of administration in patients with GCA.
7.1 Concomitant Drugs for Treatment of Adult Indications
In RA patients, population pharmacokinetic analyses did not detect any effect of methotrexate (MTX), non-steroidal anti-inflammatory drugs or corticosteroids on tocilizumab clearance. Concomitant administration of a single intravenous dose of 10 mg/kg tocilizumab with 10-25 mg MTX once weekly had no clinically significant effect on MTX exposure. Tocilizumab products have not been studied in combination with biological DMARDs such as TNF antagonists [see Dosage and Administration (2.2)].
In GCA patients, no effect of concomitant corticosteroid on tocilizumab exposure was observed.
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term animal studies have been performed to establish the carcinogenicity potential of tocilizumab products. Literature indicates that the IL-6 pathway can mediate anti-tumor responses by promoting increased immune cell surveillance of the tumor microenvironment. However, available published evidence also supports that IL-6 signaling through the IL-6 receptor may be involved in pathways that lead to tumorigenesis. The malignancy risk in humans from an antibody that disrupts signaling through the IL-6 receptor, such as tocilizumab, is presently unknown.
Fertility and reproductive performance were unaffected in male and female mice that received a murine analogue of tocilizumab administered by the intravenous route at a dose of 50 mg/kg every three days.
2.5 Recommended Dosage for Systemic Juvenile Idiopathic Arthritis
TOFIDENCE may be used as an intravenous infusion or in combination with methotrexate. Do not change a dose based solely on a single visit body weight measurement, as weight may fluctuate.
2.4 Recommended Dosage for Polyarticular Juvenile Idiopathic Arthritis
TOFIDENCE may be used as an intravenous infusion alone or in combination with methotrexate. Do not change dose based solely on a single visit body weight measurement, as weight may fluctuate.
14.4 Systemic Juvenile Idiopathic Arthritis—intravenous Administration
The efficacy of tocilizumab for the treatment of active SJIA was assessed in WA18221 (NCT00642460), a 12-week randomized, double blind, placebo-controlled, parallel group, 2-arm study. Patients treated with or without MTX, were randomized (tocilizumab:placebo = 2:1) to one of two treatment groups: 75 patients received tocilizumab infusions every two weeks at either 8 mg per kg for patients at or above 30 kg or 12 mg per kg for patients less than 30 kg and 37 were randomized to receive placebo infusions every two weeks. Corticosteroid tapering could occur from week six for patients who achieved a JIA ACR 70 response. After 12 weeks or at the time of escape, due to disease worsening, patients were treated with tocilizumab in the open-label extension phase at weight appropriate dosing.
The primary endpoint was the proportion of patients with at least 30% improvement in JIA ACR core set (JIA ACR 30 response) at Week 12 and absence of fever (no temperature at or above 37.5°C in the preceding 7 days). JIA ACR (American College of Rheumatology) responses are defined as the percentage improvement (e.g., 30%, 50%, 70%) in 3 of any 6 core outcome variables compared to baseline, with worsening in no more than 1 of the remaining variables by 30% or more. Core outcome variables consist of physician global assessment, parent per patient global assessment, number of joints with active arthritis, number of joints with limitation of movement, erythrocyte sedimentation rate (ESR), and functional ability (childhood health assessment questionnaire-CHAQ).
Primary endpoint result and JIA ACR response rates at Week 12 are shown in Table 8.
| Tocilizumab N=75 |
Placebo N=37 |
|
|---|---|---|
| Primary Endpoint: JIA ACR 30 response + absence of fever | ||
| Responders | 85% | 24% |
|
Weighted difference (95% CI) |
62 (45, 78) |
- |
| JIA ACR Response Rates at Week 12 | ||
| JIA ACR 30 | ||
| Responders | 91% | 24% |
| Weighted difference The weighted difference is the difference between the tocilizumab and Placebo response rates, adjusted for the stratification factors (weight, disease duration, background oral corticosteroid dose and background methotrexate use).
(95% CI) CI: confidence interval of the weighted difference.
|
67 (51, 83) |
- |
| JIA ACR 50 | ||
| Responders | 85% | 11% |
| Weighted difference
(95% CI) |
74 (58, 90) |
- |
| JIA ACR 70 | ||
| Responders | 71% | 8% |
| Weighted difference
(95% CI) |
63 (46, 80) |
- |
The treatment effect of tocilizumab was consistent across all components of the JIA ACR response core variables. JIA ACR scores and absence of fever responses in the open label extension were consistent with the controlled portion of the study (data available through 44 weeks).
2.7 Preparation and Administration Instructions for Intravenous Infusion
TOFIDENCE for intravenous infusion should be diluted by a healthcare professional using aseptic technique as follows:
- Use a sterile needle and syringe to prepare TOFIDENCE.
- Patients less than 30 kg: use a 50 mL infusion bag or bottle of 0.9% Sodium Chloride Injection, USP, and then follow steps 1 and 2 below.
- Patients at or above 30 kg weight: use a 100 mL infusion bag or bottle, and then follow steps 1 and 2 below.
- Step 1. Withdraw a volume of 0.9% Sodium Chloride Injection, USP, equal to the volume of the TOFIDENCE injection required for the patient’s dose from the infusion bag or bottle [see Dosage and Administration (2.2, 2.4, 2.5)].
| For Intravenous Use: Volume of TOFIDENCE Injection per kg of Body Weight | ||
|---|---|---|
| Dosage | Indication | Volume of TOFIDENCE injection per kg of body weight |
| 4 mg/kg | Adult RA | 0.2 mL/kg |
| 6 mg/kg | Adult GCA | 0.3 mL/kg |
| 8 mg/kg | Adult RA
Adult COVID-19 SJIA and PJIA (greater than or equal to 30 kg of body weight) |
0.4 mL/kg |
| 10 mg/kg | PJIA (less than 30 kg of body weight) | 0.5 mL/kg |
| 12 mg/kg | SJIA (less than 30 kg of body weight) | 0.6 mL/kg |
- Step 2. Withdraw the amount of TOFIDENCE for intravenous infusion from the vial(s) and add slowly into the 0.9% Sodium Chloride Injection, USP infusion bag or bottle. To mix the solution, gently invert the bag to avoid foaming.
- The fully diluted TOFIDENCE solutions for infusion using 0.9% Sodium Chloride Injection, USP may be stored refrigerated at 36°F to 46°F (2°C to 8°C) for up to 24 hours or room temperature at 68°F to 77°F (20°C to 25°C) for up to 12 hours and should be protected from light.
- TOFIDENCE solutions do not contain preservatives; therefore, unused product remaining in the vials should not be used.
- Allow the fully diluted TOFIDENCE solution to reach room temperature prior to infusion.
- The infusion should be administered over 60 minutes, and must be administered with an infusion set. Do not administer as an intravenous push or bolus.
- TOFIDENCE should not be infused concomitantly in the same intravenous line with other drugs. No physical or biochemical compatibility studies have been conducted to evaluate the co-administration of TOFIDENCE with other drugs.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If particulates and discolorations are noted, the product should not be used.
- Fully diluted TOFIDENCE solutions are compatible with infusion bags and/or infusion sets with the following materials: polypropylene, polyethylene, polyolefin, polyvinyl chloride, polyethersulfone, polyurethane, nylon and stainless steel.
14.3 Polyarticular Juvenile Idiopathic Arthritis—intravenous Administration
The efficacy of tocilizumab was assessed in a three-part study, WA19977 (NCT00988221), including an open-label extension in children 2 to 17 years of age with active polyarticular juvenile idiopathic arthritis (PJIA), who had an inadequate response to methotrexate or inability to tolerate methotrexate. Patients had at least 6 months of active disease (mean disease duration of 4.2 ± 3.7 years), with at least five joints with active arthritis (swollen or limitation of movement accompanied by pain and/or tenderness) and/or at least 3 active joints having limitation of motion (mean, 20 ± 14 active joints). The patients treated had subtypes of JIA that at disease onset included Rheumatoid Factor Positive or Negative Polyarticular JIA, or Extended Oligoarticular JIA. Treatment with a stable dose of methotrexate was permitted but was not required during the study. Concurrent use of disease modifying antirheumatic drugs (DMARDs), other than methotrexate, or other biologics (e.g., TNF antagonists or T cell costimulation modulator) were not permitted in the study.
Part I consisted of a 16-week active tocilizumab treatment lead-in period (n=188) followed by Part II, a 24-week randomized double-blind placebo-controlled withdrawal period, followed by Part III, a 64-week open-label period. Eligible patients weighing at or above 30 kg received tocilizumab at 8 mg/kg intravenously once every four weeks. Patients weighing less than 30 kg were randomized 1:1 to receive either tocilizumab 8 mg/kg or 10 mg/kg intravenously every four weeks. At the conclusion of the open-label Part I, 91% of patients taking background MTX in addition to tocilizumab and 83% of patients on tocilizumab monotherapy achieved an ACR 30 response at week 16 compared to baseline and entered the blinded withdrawal period (Part II) of the study. The proportions of patients with JIA ACR 50/70 responses in Part I were 84.0%, and 64%, respectively for patients taking background MTX in addition to tocilizumab and 80% and 55% respectively for patients on tocilizumab monotherapy.
In Part II, patients (ITT, n=163) were randomized to tocilizumab (same dose received in Part I) or placebo in a 1:1 ratio that was stratified by concurrent methotrexate use and concurrent corticosteroid use. Each patient continued in Part II of the study until Week 40 or until the patient satisfied JIA ACR 30 flare criteria (relative to Week 16) and qualified for escape.
The primary endpoint was the proportion of patients with a JIA ACR 30 flare at week 40 relative to week 16. JIA ACR 30 flare was defined as 3 or more of the 6 core outcome variables worsening by at least 30% with no more than 1 of the remaining variables improving by more than 30% relative to Week 16.
Tocilizumab treated patients experienced significantly fewer disease flares compared to placebo-treated patients (26% [21/82] versus 48% [39/81]; adjusted difference in proportions -21%, 95% CI: -35%, -8%).
During the withdrawal phase (Part II), more patients treated with tocilizumab showed JIA ACR 30/50/70 responses at Week 40 compared to patients withdrawn to placebo.
6.5 Clinical Trials Experience in Covid 19 Patients Treated With Intravenous Tocilizumab (tocilizumab Iv)
The safety of tocilizumab in hospitalized COVID-19 patients was evaluated in a pooled safety population that includes patients enrolled in EMPACTA, COVACTA, AND REMDACTA. The analysis of adverse reactions included a total of 974 patients exposed to tocilizumab. Patients received a single, 60-minute infusion of intravenous tocilizumab 8 mg/kg (maximum dose of 800 mg). If clinical signs or symptoms worsened or did not improve, one additional dose of tocilizumab 8 mg/kg could be administered between 8- 24 hours after the initial dose.
Adverse reactions summarized in Table 3 occurred in at least 3% of tocilizumab -treated patients and more commonly than in patients on placebo in the pooled safety population.
| Adverse Reaction | Tocilizumab 8mg per kg |
Placebo |
|---|---|---|
| N = 974 (%) |
N = 483 (%) |
|
| Hepatic Transaminases increased | 10% | 8% |
| Constipation | 9% | 8% |
| Urinary tract infection | 5% | 4% |
| Hypertension | 4% | 1% |
| Hypokalaemia | 4% | 3% |
| Anxiety | 4% | 2% |
| Diarrhea | 4% | 2% |
| Insomnia | 4% | 3% |
| Nausea | 3% | 2% |
In the pooled safety population, the rates of infection/serious infection events were 30%/19% in patients receiving tocilizumab versus 32%/23% receiving placebo.
6.1 Clinical Trials Experience in Rheumatoid Arthritis Patients Treated With Intravenous Tocilizumab (tocilizumab Iv)
The tocilizumab-IV data in rheumatoid arthritis (RA) includes 5 double-blind, controlled, multicenter studies. In these studies, patients received doses of tocilizumab-IV 8 mg per kg monotherapy (288 patients), tocilizumab-IV 8 mg per kg in combination with DMARDs (including methotrexate) (1582 patients), or tocilizumab-IV 4 mg per kg in combination with methotrexate (774 patients).
The all exposure population includes all patients in registration studies who received at least one dose of tocilizumab-IV. Of the 4009 patients in this population, 3577 received treatment for at least 6 months, 3309 for at least one year; 2954 received treatment for at least 2 years and 2189 for 3 years.
All patients in these studies had moderately to severely active rheumatoid arthritis. The study population had a mean age of 52 years, 82% were female and 74% were Caucasian.
The most common serious adverse reactions were serious infections [see Warnings and Precautions (5.1)]. The most commonly reported adverse reactions in controlled studies up to 24 weeks (occurring in at least 5% of patients treated with tocilizumab-IV monotherapy or in combination with DMARDs) were upper respiratory tract infections, nasopharyngitis, headache, hypertension and increased ALT.
The proportion of patients who discontinued treatment due to any adverse reactions during the double-blind, placebo-controlled studies was 5% for patients taking tocilizumab-IV and 3% for placebo-treated patients. The most common adverse reactions that required discontinuation of tocilizumab-IV were increased hepatic transaminase values (per protocol requirement) and serious infections.
6.2 Clinical Trials Experience in Giant Cell Arteritis Patients Treated With Intravenous Tocilizumab (tocilizumab Iv)
The safety of tocilizumab-IV was studied in an open label PK-PD and safety study in 24 patients with GCA who were in remission on tocilizumab-IV at time of enrollment. Patients received tocilizumab 7 mg/kg every 4 weeks for 20 weeks, followed by 6 mg/kg every 4 weeks for 20 weeks. The total patient years exposure to treatment was 17.5 years.
The safety of tocilizumab by another route of administration has been studied in one Phase III study with 251 GCA patients. The total patient years duration was 138.5 patient years during the 12-month double blind, placebo-controlled phase of the study. The overall safety profile observed was generally consistent with the known safety profile of tocilizumab. There was an overall higher incidence of infections in GCA patients relative to RA patients.
The overall safety profile observed for tocilizumab administered intravenously in GCA patients was consistent with the known safety profile of tocilizumab.
6.4 Clinical Trials Experience in Systemic Juvenile Idiopathic Arthritis Patients Treated With Intravenous Tocilizumab (tocilizumab Iv)
The data described below reflect exposure to tocilizumab-IV in one randomized, double-blind, placebo-controlled trial of 112 pediatric patients with SJIA 2 to 17 years of age who had an inadequate clinical response to nonsteroidal anti-inflammatory drugs (NSAIDs) or corticosteroids due to toxicity or lack of efficacy. At baseline, approximately half of the patients were taking 0.3 mg/kg/day corticosteroids or more, and almost 70% were taking methotrexate. The trial included a 12 week controlled phase followed by an open-label extension. In the 12 week double-blind, controlled portion of the clinical study 75 patients received treatment with tocilizumab-IV (8 or 12 mg per kg based upon body weight). After 12 weeks or at the time of escape, due to disease worsening, patients were treated with tocilizumab-IV in the open-label extension phase.
The most common adverse events (at least 5%) seen in tocilizumab-IV treated patients in the 12 week controlled portion of the study were: upper respiratory tract infection, headache, nasopharyngitis and diarrhea.
6.3 Clinical Trials Experience in Polyarticular Juvenile Idiopathic Arthritis Patients Treated With Intravenous Tocilizumab (tocilizumab Iv)
The safety of tocilizumab-IV was studied in 188 pediatric patients 2 to 17 years of age with PJIA who had an inadequate clinical response or were intolerant to methotrexate. The total patient exposure in the tocilizumab-IV all exposure population (defined as patients who received at least one dose of tocilizumab-IV) was 184.4 patient years. At baseline, approximately half of the patients were taking oral corticosteroids and almost 80% were taking methotrexate. In general, the types of adverse drug reactions in patients with PJIA were consistent with those seen in RA and SJIA patients [see Adverse Reactions (6.1 and 6.4)].
Structured Label Content
Section 42229-5 (42229-5)
Not Recommended for Concomitant Use with Biological DMARDs
Tocilizumab products have not been studied in combination with biological DMARDs such as TNF antagonists, IL-1R antagonists, anti-CD20 monoclonal antibodies and selective co-stimulation modulators because of the possibility of increased immunosuppression and increased risk of infection. Avoid using TOFIDENCE with biological DMARDs.
Section 42231-1 (42231-1)
| Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: 07/2025 | |||
|
Medication Guide
TOFIDENCE® (TOE-FIH-DENCE) (tocilizumab-bavi) injection for intravenous use |
||||
|
What is the most important information I should know about TOFIDENCE?
Your healthcare provider should monitor you closely for signs and symptoms of TB during and after treatment with TOFIDENCE.
|
||||
|
|
|
||
|
||||
|
|
|||
You should not receive TOFIDENCE if your neutrophil or platelet counts are too low or your liver function tests are too high. Your healthcare provider may stop your TOFIDENCE treatment for a period of time or change your dose of medicine if needed because of changes in these blood test results.
|
||||
|
What is TOFIDENCE?
TOFIDENCE is a prescription medicine called an Interleukin-6 (IL-6) receptor antagonist. TOFIDENCE is used:
|
||||
| Do not take TOFIDENCE: if you are allergic to tocilizumab products, or any of the ingredients in TOFIDENCE. See the end of this Medication Guide for a complete list of ingredients in TOFIDENCE. | ||||
Before you receive TOFIDENCE, tell your healthcare provider about all of your medical conditions, including if you:
|
||||
|
Tell your healthcare provider about all of the medicines you take, including prescription, over-the-counter medicines, vitamins and herbal supplements. TOFIDENCE and other medicines may affect each other causing side effects. Especially tell your healthcare provider if you take:
|
||||
|
How will I receive TOFIDENCE?
Into a vein (IV or intravenous infusion) for Rheumatoid Arthritis, Giant Cell Arteritis, PJIA, SJIA, or COVID-19:
|
||||
|
What are the possible side effects with TOFIDENCE?
|
||||
|
|
|
||
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Organon USA LLC, a subsidiary of Organon & Co., at 1-844-674-3200. |
||||
|
General information about the safe and effective use of TOFIDENCE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not give TOFIDENCE to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TOFIDENCE that is written for health professionals. |
||||
|
What are the ingredients in TOFIDENCE?
Active ingredient: tocilizumab-bavi. Inactive ingredients of Intravenous TOFIDENCE: arginine hydrochloride, histidine, L-histidine hydrochloride monohydrate,polysorbate 80, sucrose, and water for Injection. Manufactured by: Organon USA LLC, a subsidiary of ORGANON & Co., Jersey City, NJ 07302, USA U.S. License No. 2331 © 2025 Organon group of companies. All rights reserved. For more information, go to www.tofidence.com or call 1-844-674-3200 . usmg-og9401-i-2507r000 |
||||
Section 43683-2 (43683-2)
Section 44425-7 (44425-7)
Storage and Handling: Do not use beyond expiration date on the container or package. TOFIDENCE must be refrigerated at 36°F to 46°F (2°C to 8°C). Do not freeze. Protect the vials from light by storage in the original package until time of use.
10 Overdosage (10 OVERDOSAGE)
There are limited data available on overdoses with tocilizumab products. One case of accidental overdose was reported with intravenous tocilizumab in which a patient with multiple myeloma received a dose of 40 mg per kg. No adverse drug reactions were observed. No serious adverse drug reactions were observed in healthy volunteers who received single doses of up to 28 mg per kg, although all 5 patients at the highest dose of 28 mg per kg developed dose-limiting neutropenia.
In case of an overdose, it is recommended that the patient be monitored for signs and symptoms of adverse reactions. Patients who develop adverse reactions should receive appropriate symptomatic treatment.
11 Description (11 DESCRIPTION)
Tocilizumab-bavi is a recombinant humanized anti-human interleukin 6 (IL-6) receptor monoclonal antibody of the immunoglobulin IgG1κ (gamma 1, kappa) subclass with a typical H2L2 polypeptide structure. Each light chain and heavy chain consists of 214 and 448 amino acids, respectively. The four polypeptide chains are linked intra- and inter-molecularly by disulfide bonds. Tocilizumab-bavi has a molecular weight of approximately 148 kDa. The antibody is produced in mammalian (Chinese hamster ovary) cells.
5.9 Vaccinations
Avoid use of live vaccines concurrently with TOFIDENCE as clinical safety has not been established. No data are available on the secondary transmission of infection from persons receiving live vaccines to patients receiving tocilizumab products.
No data are available on the effectiveness of vaccination in patients receiving tocilizumab products. Because IL-6 inhibition may interfere with the normal immune response to new antigens, it is recommended that all patients, particularly pediatric or elderly patients, if possible, be brought up to date with all immunizations in agreement with current immunization guidelines prior to initiating TOFIDENCE therapy. The interval between live vaccinations and initiation of TOFIDENCE therapy should be in accordance with current vaccination guidelines regarding immunosuppressive agents.
7.3 Live Vaccines
Avoid use of live vaccines concurrently with TOFIDENCE [see Warnings and Precautions (5.9)].
8.4 Pediatric Use
TOFIDENCE by intravenous use is indicated for the treatment of pediatric patients with:
- Active systemic juvenile idiopathic arthritis in patients 2 years of age and older
- Active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older
The safety and effectiveness of TOFIDENCE in pediatric patients with conditions other than PJIA or SJIA have not been established. The safety and effectiveness in pediatric patients below the age of 2 have not been established in PJIA or SJIA.
8.5 Geriatric Use
Of the 2644 patients who received tocilizumab in Studies I to V [see Clinical Studies (14)], a total of 435 rheumatoid arthritis patients were 65 years of age and older, including 50 patients 75 years and older. The frequency of serious infection among tocilizumab treated subjects 65 years of age and older was higher than those under the age of 65. As there is a higher incidence of infections in the elderly population in general, caution should be used when treating the elderly.
In the EMPACTA, COVACTA, and REMDACTA studies, of the 974 COVID-19 patients in the tocilizumab arm, 375 (39%) were 65 years of age or older. No overall differences in safety or effectiveness of tocilizumab were observed between patients 65 years of age and older and those under the age of 65 years of age in these studies [see Adverse Reactions (6.5) and Clinical Studies (14.5)].
In the RECOVERY study, of the 2022 COVID-19 patients in the tocilizumab arm, 930 (46%) were 65 years of age or older. No overall differences in effectiveness of tocilizumab were observed between patients 65 years of age and older and those under the age 65 years of age in this study [see Clinical Studies (14.5)].
5.3 Hepatotoxicity
Serious cases of hepatic injury have been observed in patients taking intravenous tocilizumab products. Some of these cases have resulted in liver transplant or death. Time to onset for cases ranged from months to years after treatment initiation with tocilizumab products. While most cases presented with marked elevations of transaminases (> 5 times ULN), some cases presented with signs or symptoms of liver dysfunction and only mildly elevated transaminases.
During randomized controlled studies, treatment with tocilizumab was associated with a higher incidence of transaminase elevations [see Adverse Reactions (6.1, 6.3, 6.4)]. Increased frequency and magnitude of these elevations was observed when potentially hepatotoxic drugs (e.g., MTX) were used in combination with tocilizumab.
For RA and GCA patients, obtain a liver test panel (serum alanine aminotransferase [ALT], aspartate aminotransferase [AST], alkaline phosphatase, and total bilirubin) before initiating TOFIDENCE, every 4 to 8 weeks after start of therapy for the first 6 months of treatment and every 3 months thereafter. It is not recommended to initiate TOFIDENCE treatment in RA or GCA patients with elevated transaminases ALT or AST greater than 1.5x ULN. In patients who develop elevated ALT or AST greater than 5x ULN, discontinue TOFIDENCE. For recommended modifications based upon increase in transaminases [see Dosage and Administration (2.8)].
Patients hospitalized with COVID-19 may have elevated ALT or AST levels. Multi-organ failure with involvement of the liver is recognized as a complication of severe COVID-19. The decision to administer TOFIDENCE should balance the potential benefit of treating COVID-19 against the potential risks of acute treatment with TOFIDENCE. It is not recommended to initiate TOFIDENCE treatment in COVID-19 patients with elevated ALT or AST above 10 x ULN. Monitor ALT and AST during treatment.
Measure liver tests promptly in patients who report symptoms that may indicate liver injury, such as fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice. In this clinical context, if the patient is found to have abnormal liver tests (e.g., ALT greater than three times the upper limit of the reference range, serum total bilirubin greater than two times the upper limit of the reference range), TOFIDENCE treatment should be interrupted and investigation done to establish the probable cause. TOFIDENCE should only be restarted in patients with another explanation for the liver test abnormalities after normalization of the liver tests.
A similar pattern of liver enzyme elevation is noted with tocilizumab products treatment in the PJIA and SJIA populations. Monitor liver test panel at the time of the second administration and thereafter every 4 to 8 weeks for PJIA and every 2 to 4 weeks for SJIA.
4 Contraindications (4 CONTRAINDICATIONS)
TOFIDENCE is contraindicated in patients with known hypersensitivity to tocilizumab products [see Warnings and Precautions (5.6)].
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following serious adverse reactions are described elsewhere in labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Gastrointestinal Perforations [see Warnings and Precautions (5.2)]
- Laboratory Parameters [see Warnings and Precautions (5.4)]
- Immunosuppression [see Warnings and Precautions (5.5)]
- Hypersensitivity Reactions, Including Anaphylaxis [see Warnings and Precautions (5.6)]
- Demyelinating Disorders [see Warnings and Precautions (5.7)]
- Active Hepatic Disease and Hepatic Impairment [see Warnings and Precautions (5.8)]
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not predict the rates observed in a broader patient population in clinical practice.
8.7 Renal Impairment
No dose adjustment is required in patients with mild or moderate renal impairment. Tocilizumab products have not been studied in patients with severe renal impairment [see Clinical Pharmacology (12.3)].
12.2 Pharmacodynamics
In clinical studies in RA patients with the 4 mg per kg and 8 mg per kg intravenous doses decreases in levels of C-reactive protein (CRP) to within normal ranges were seen as early as week 2. Changes in pharmacodynamic parameters were observed (i.e., decreases in rheumatoid factor, erythrocyte sedimentation rate (ESR), serum amyloid A, fibrinogen and increases in hemoglobin) with doses, however the greatest improvements were observed with 8 mg per kg tocilizumab. Pharmacodynamic changes were also observed to occur after tocilizumab administration in GCA, PJIA, and SJIA patients (decreases in CRP, ESR, and increases in hemoglobin). The relationship between these pharmacodynamic findings and clinical efficacy is not known.
In healthy subjects administered tocilizumab in doses from 2 to 28 mg per kg intravenously, absolute neutrophil counts decreased to the nadir 3 to 5 days following tocilizumab administration. Thereafter, neutrophils recovered towards baseline in a dose dependent manner. Rheumatoid arthritis and GCA patients demonstrated a similar pattern of absolute neutrophil counts following tocilizumab administration [see Warnings and Precautions (5.4)].
12.3 Pharmacokinetics
PK of tocilizumab is characterized by nonlinear elimination which is a combination of linear clearance and Michaelis-Menten elimination. The nonlinear part of tocilizumab elimination leads to an increase in exposure that is more than dose-proportional. The pharmacokinetic parameters of tocilizumab do not change with time. Due to the dependence of total clearance on tocilizumab serum concentrations, the half-life of tocilizumab is also concentration-dependent and varies depending on the serum concentration level. Population pharmacokinetic analyses in any patient population tested so far indicate no relationship between apparent clearance and the presence of anti-drug antibodies.
5.5 Immunosuppression
The impact of treatment with tocilizumab products on the development of malignancies is not known but malignancies were observed in clinical studies [see Adverse Reactions (6.1)]. TOFIDENCE is an immunosuppressant, and treatment with immunosuppressants may result in an increased risk of malignancies.
5.1 Serious Infections
Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, protozoal, or other opportunistic pathogens have been reported in patients receiving immunosuppressive agents including tocilizumab products. The most common serious infections included pneumonia, urinary tract infection, cellulitis, herpes zoster, gastroenteritis, diverticulitis, sepsis and bacterial arthritis [see Adverse Reactions (6.1)]. Among opportunistic infections, tuberculosis, cryptococcus, aspergillosis, candidiasis, and pneumocystosis were reported with tocilizumab products. Other serious infections, not reported in clinical studies, may also occur (e.g., histoplasmosis, coccidioidomycosis, listeriosis). Patients have presented with disseminated rather than localized disease, and were often taking concomitant immunosuppressants such as methotrexate or corticosteroids which in addition to rheumatoid arthritis may predispose them to infections.
Do not administer TOFIDENCE in patients with an active infection, including localized infections. The risks and benefits of treatment should be considered prior to initiating TOFIDENCE in patients:
- with chronic or recurrent infection;
- who have been exposed to tuberculosis;
- with a history of serious or an opportunistic infection;
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
- with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with TOFIDENCE, as signs and symptoms of acute inflammation may be lessened due to suppression of the acute phase reactants [see Dosage and Administration (2.1), Adverse Reactions (6.1), and Patient Counseling Information (17)].
Hold TOFIDENCE if a patient develops a serious infection, an opportunistic infection, or sepsis. A patient who develops a new infection during treatment with TOFIDENCE should undergo a prompt and complete diagnostic workup appropriate for an immunocompromised patient, initiate appropriate antimicrobial therapy, and closely monitor the patient.
8.6 Hepatic Impairment
The safety and efficacy of tocilizumab products have not been studied in patients with hepatic impairment, including patients with positive HBV and HCV serology [see Warnings and Precautions (5.8)].
1 Indications and Usage (1 INDICATIONS AND USAGE)
TOFIDENCE® (tocilizumab-bavi) is an interleukin-6 (IL-6) receptor antagonist indicated for treatment of:
Rheumatoid Arthritis (RA) (1.1)
- Adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more Disease-Modifying Anti-Rheumatic Drugs (DMARDs).
Giant Cell Arteritis (GCA) (1.2)
- Adult patients with giant cell arteritis.
Polyarticular Juvenile Idiopathic Arthritis (PJIA) (1.3)
- Patients 2 years of age and older with active polyarticular juvenile idiopathic arthritis.
Systemic Juvenile Idiopathic Arthritis (SJIA) (1.4)
- Patients 2 years of age and older with active systemic juvenile idiopathic arthritis.
Coronavirus Disease 2019 (COVID-19) (1.5)
- Hospitalized adult patients with coronavirus disease 2019 (COVID-19) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
12.1 Mechanism of Action
Tocilizumab products bind to both soluble and membrane-bound IL-6 receptors (sIL-6R and mIL-6R), and have been shown to inhibit IL-6-mediated signaling through these receptors. IL-6 is a pleiotropic pro-inflammatory cytokine produced by a variety of cell types including T- and B-cells, lymphocytes, monocytes and fibroblasts. IL-6 has been shown to be involved in diverse physiological processes such as T-cell activation, induction of immunoglobulin secretion, initiation of hepatic acute phase protein synthesis, and stimulation of hematopoietic precursor cell proliferation and differentiation. IL-6 is also produced by synovial and endothelial cells leading to local production of IL-6 in joints affected by inflammatory processes such as rheumatoid arthritis.
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Serious Infections – do not administer TOFIDENCE during an active infection, including localized infections. If a serious infection develops, interrupt TOFIDENCE until the infection is controlled. (5.1)
- Gastrointestinal (GI) perforation—use with caution in patients who may be at increased risk. (5.2)
- Hepatotoxicity- Monitor patients for signs and symptoms of hepatic injury. Modify or discontinue TOFIDENCE if abnormal liver tests persist or worsen or if clinical signs and symptoms of liver disease develop. (2.8, 5.3)
- Laboratory monitoring—recommended due to potential consequences of treatment-related changes in neutrophils, platelets, lipids, and liver function tests. (2.8, 5.4)
- Hypersensitivity reactions, including anaphylaxis and death and serious cutaneous reactions including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) – discontinue TOFIDENCE, treat promptly, and monitor until reaction resolves. (5.6)
- Live vaccines—Avoid use with TOFIDENCE. (5.9, 7.3)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
For RA, pJIA and sJIA, TOFIDENCE may be used alone or in combination with methotrexate; and in RA, other non-biologic DMARDs may be used. (2)
General Administration and Dosing Information (2.1)
- RA, GCA, PJIA and SJIA - It is recommended that TOFIDENCE not be initiated in patients with an absolute neutrophil count (ANC) below 2000 per mm3, platelet count below 100,000 per mm3, or ALT or AST above 1.5 times the upper limit of normal (ULN). (5.3, 5.4)
- COVID-19 - It is recommended that TOFIDENCE not be initiated in patients with an absolute neutrophil count (ANC) below 1000 per mm3 , platelet count below 50,000 mm3 , or ALT or AST above 10 times ULN. (5.3, 5.4)
- In RA or COVID-19 patients, TOFIDENCE doses exceeding 800 mg per infusion are not recommended. (2.2, 12.3)
- In GCA patients, TOFIDENCE doses exceeding 600 mg per infusion are not recommended. (2.3, 12.3)
Rheumatoid Arthritis (2.2)
Recommended Adult Intravenous Dosage:
When used in combination with non-biologic DMARDs or as monotherapy the recommended starting dose is 4 mg per kg every 4 weeks followed by an increase to 8 mg per kg every 4 weeks based on clinical response.
Giant Cell Arteritis (2.3)
Recommended Adult Intravenous Dosage:
The recommended dose is 6 mg per kg every 4 weeks in combination with a tapering course of glucocorticoids. TOFIDENCE can be used alone following discontinuation of glucocorticoids.
Polyarticular Juvenile Idiopathic Arthritis (2.4)
| Recommended Intravenous PJIA Dosage Every 4 Weeks | |
|---|---|
| Patients less than 30 kg weight | 10 mg per kg |
| Patients at or above 30 kg weight | 8 mg per kg |
Systemic Juvenile Idiopathic Arthritis (2.5)
| Recommended Intravenous SJIA Dosage Every 2 Weeks | |
|---|---|
| Patients less than 30 kg weight | 12 mg per kg |
| Patients at or above 30 kg weight | 8 mg per kg |
Coronavirus Disease 2019 (2.6)
The recommended dosage of TOFIDENCE for adult patients with COVID-19 is 8 mg per kg administered by a 60-minute intravenous infusion.
Administration of Intravenous Formulation (2.7)
- For patients with RA, GCA, COVID-19, PJIA, and SJIA patients at or above 30 kg, dilute to 100 mL in 0.9% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique.
- For PJIA and SJIA patients less than 30 kg, dilute to 50 mL in 0.9% Sodium Chloride Injection, USP for intravenous infusion using aseptic technique.
- Administer as a single intravenous drip infusion over 1 hour; do not administer as bolus or push.
Dose Modifications (2.8)
- Recommended for management of certain dose-related laboratory changes including elevated liver enzymes, neutropenia, and thrombocytopenia.
5.7 Demyelinating Disorders
The impact of treatment with tocilizumab products on demyelinating disorders is not known, but multiple sclerosis and chronic inflammatory demyelinating polyneuropathy were reported rarely in RA clinical studies. Monitor patients for signs and symptoms potentially indicative of demyelinating disorders. Prescribers should exercise caution in considering the use of TOFIDENCE in patients with preexisting or recent onset demyelinating disorders.
9 Drug Abuse and Dependence (9 DRUG ABUSE AND DEPENDENCE)
No studies on the potential for tocilizumab products to cause dependence have been performed. However, there is no evidence from the available data that tocilizumab products treatment results in dependence.
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Intravenous Infusion
Injection: 80 mg/4 mL (20 mg/mL), 200 mg/10 mL (20 mg/mL), 400 mg/20 mL (20 mg/mL) in single-dose vials for further dilution prior to intravenous infusion (3)
6.6 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of tocilizumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hypersensitivity Reactions: Fatal anaphylaxis, Stevens-Johnson Syndrome, Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) [see Warnings and Precautions (5.6)]
- Pancreatitis
- Drug-induced liver injury, Hepatitis, Hepatic failure, Jaundice [see Warnings and Precautions (5.3)]
1.1 Rheumatoid Arthritis (ra) (1.1 Rheumatoid Arthritis (RA))
TOFIDENCE (tocilizumab-bavi) is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more Disease-Modifying Anti-Rheumatic Drugs (DMARDs).
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
1.2 Giant Cell Arteritis (gca) (1.2 Giant Cell Arteritis (GCA))
TOFIDENCE® (tocilizumab-bavi) is indicated for the treatment of giant cell arteritis (GCA) in adult patients.
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
5.2 Gastrointestinal Perforations
Events of gastrointestinal perforation have been reported in clinical trials, primarily as complications of diverticulitis in patients treated with tocilizumab. Use TOFIDENCE with caution in patients who may be at increased risk for gastrointestinal perforation. Promptly evaluate patients presenting with fever, new onset abdominal symptoms, and a change in bowel habits for early identification of gastrointestinal perforation [see Adverse Reactions (6.1)].
Warning: Risk of Serious Infections (WARNING: RISK OF SERIOUS INFECTIONS)
Patients treated with tocilizumab products including TOFIDENCE are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
If a serious infection develops, interrupt TOFIDENCE until the infection is controlled.
Reported infections include:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients, except those with COVID-19, should be tested for latent tuberculosis before TOFIDENCE use and during therapy. Treatment for latent infection should be initiated prior to TOFIDENCE use.
- Invasive fungal infections, including candidiasis, aspergillosis, and pneumocystis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral and other infections due to opportunistic pathogens.
The risks and benefits of treatment with TOFIDENCE should be carefully considered prior to initiating therapy in patients with chronic or recurrent infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with TOFIDENCE, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1)].
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
TOFIDENCE (tocilizumab-bavi) injection is a preservative-free, sterile, clear to opalescent, colorless to light yellow solution. TOFIDENCE is supplied as 80 mg/4 mL (NDC 78206-200-01), 200 mg/10 mL (NDC 78206-201-01), and 400 mg/20 mL (NDC 78206-202-01) individually packaged 20 mg/mL single-dose vials for further dilution prior to intravenous infusion.
1.5 Coronavirus Disease 2019 (covid 19) (1.5 Coronavirus Disease 2019 (COVID-19))
TOFIDENCE® (tocilizumab-bavi) is indicated for the treatment of coronavirus disease 2019 (COVID-19) in hospitalized adult patients who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
2.6 Coronavirus Disease 2019 (covid 19) (2.6 Coronavirus Disease 2019 (COVID-19))
Administer TOFIDENCE by intravenous infusion only.
The recommended dosage of TOFIDENCE for treatment of adult patients with COVID-19 is 8 mg per kg administered as a single 60-minute intravenous infusion. If clinical signs or symptoms worsen or do not improve after the first dose, one additional infusion of TOFIDENCE may be administered at least 8 hours after the initial infusion.
- Doses exceeding 800 mg per infusion are not recommended in patients with COVID-19.
7.2 Interactions With Cyp450 Substrates (7.2 Interactions with CYP450 Substrates)
Cytochrome P450s in the liver are down-regulated by infection and inflammation stimuli including cytokines such as IL-6. Inhibition of IL-6 signaling in RA patients treated with tocilizumab products may restore CYP450 activities to higher levels than those in the absence of tocilizumab products leading to increased metabolism of drugs that are CYP450 substrates. In vitro studies showed that tocilizumab has the potential to affect expression of multiple CYP enzymes including CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. Its effect on CYP2C8 or transporters is unknown. In vivo studies with omeprazole, metabolized by CYP2C19 and CYP3A4, and simvastatin, metabolized by CYP3A4, showed up to a 28% and 57% decrease in exposure one week following a single dose of tocilizumab, respectively. The effect of tocilizumab products on CYP enzymes may be clinically relevant for CYP450 substrates with narrow therapeutic index, where the dose is individually adjusted. Upon initiation or discontinuation of TOFIDENCE, in patients being treated with these types of medicinal products, perform therapeutic monitoring of effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) and the individual dose of the medicinal product adjusted as needed. Exercise caution when coadministering TOFIDENCE with CYP3A4 substrate drugs where decrease in effectiveness is undesirable, e.g., oral contraceptives, lovastatin, atorvastatin, etc. The effect of tocilizumab products on CYP450 enzyme activity may persist for several weeks after stopping therapy [see Clinical Pharmacology (12.3)].
14.5 Covid 19 Intravenous Administration (14.5 COVID-19 - Intravenous Administration)
The efficacy of tocilizumab for the treatment of COVID-19 was based on RECOVERY (NCT04381936), a randomized, controlled, open-label, platform study, and supported by the results from EMPACTA (NCT04372186), a randomized, double-blind, placebo-controlled study. Results of two other randomized, double-blind, placebo-controlled studies, COVACTA (NCT04320615) and REMDACTA (NCT04409262), which evaluated the efficacy of tocilizumab for the treatment of COVID-19 are also summarized.
2.2 Recommended Dosage for Rheumatoid Arthritis
TOFIDENCE may be used as monotherapy or concomitantly with methotrexate or other non-biologic DMARDs as an intravenous infusion.
Principal Display Panel 80 Mg/4 Ml Vial Carton (PRINCIPAL DISPLAY PANEL - 80 mg/4 mL Vial Carton)
Rx Only
NDC 78206-200-01
Tofidence®
(tocilizumab-bavi)
Injection
80 mg/4 mL
(20 mg/mL)
For Intravenous Infusion
after dilution
Single-Dose Vial
Discard unused portion.
ORGANON
1.4 Systemic Juvenile Idiopathic Arthritis (sjia) (1.4 Systemic Juvenile Idiopathic Arthritis (SJIA))
TOFIDENCE® (tocilizumab-bavi) is indicated for the treatment of active systemic juvenile idiopathic arthritis in patients 2 years of age and older.
5.8 Active Hepatic Disease and Hepatic Impairment
Treatment with TOFIDENCE is not recommended in patients with active hepatic disease or hepatic impairment [see Adverse Reactions (6.1), Use in Specific Populations (8.6)].
Principal Display Panel 200 Mg/10 Ml Vial Carton (PRINCIPAL DISPLAY PANEL - 200 mg/10 mL Vial Carton)
Rx Only
NDC 78206-201-01
Tofidence®
(tocilizumab-bavi)
Injection
200 mg/10 mL
(20 mg/mL)
For Intravenous Infusion
after dilution
Single-Dose Vial
Discard unused portion.
ORGANON
Principal Display Panel 400 Mg/20 Ml Vial Carton (PRINCIPAL DISPLAY PANEL - 400 mg/20 mL Vial Carton)
Rx Only
NDC 78206-202-01
Tofidence®
(tocilizumab-bavi)
Injection
400 mg/20 mL
(20 mg/mL)
For Intravenous Infusion
after dilution
Single-Dose Vial
Discard unused portion.
ORGANON
5.6 Hypersensitivity Reactions, Including Anaphylaxis
Hypersensitivity reactions, including anaphylaxis, have been reported in association with tocilizumab products and anaphylactic events with a fatal outcome have been reported with intravenous infusion of tocilizumab products. Anaphylaxis and other hypersensitivity reactions that required treatment discontinuation were reported in 0.1% (3 out of 2644) of patients in the 6-month controlled trials of intravenous tocilizumab and 0.2% (8 out of 4009) of patients in the intravenous all-exposure RA population. In the SJIA controlled trial with intravenous tocilizumab, 1 out of 112 patients (0.9%) experienced hypersensitivity reactions that required treatment discontinuation. In the PJIA controlled trial with intravenous tocilizumab 0 out of 188 patients (0%) in the tocilizumab all-exposure population experienced hypersensitivity reactions that required treatment discontinuation. Reactions that required treatment discontinuation included generalized erythema, rash, and urticaria.
In the postmarketing setting, events of hypersensitivity reactions, including anaphylaxis and death have occurred in patients treated with a range of doses of intravenous tocilizumab products, with or without concomitant therapies. Events have occurred in patients who received premedication. Hypersensitivity, including anaphylaxis events, have occurred both with and without previous hypersensitivity reactions and as early as the first infusion of tocilizumab products [see Adverse Reactions (6.6)]. In addition, serious cutaneous reactions, including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), have been reported in patients with autoinflammatory conditions treated with tocilizumab products.
TOFIDENCE for intravenous use should only be infused by a healthcare professional with appropriate medical support to manage anaphylaxis. If a hypersensitivity reaction occurs, immediately discontinue TOFIDENCE, treat promptly and monitor until signs and symptoms resolve.
1.3 Polyarticular Juvenile Idiopathic Arthritis (pjia) (1.3 Polyarticular Juvenile Idiopathic Arthritis (PJIA))
TOFIDENCE® (tocilizumab-bavi) is indicated for the treatment of active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older.
14.1 Rheumatoid Arthritis—intravenous Administration (14.1 Rheumatoid Arthritis—Intravenous Administration)
The efficacy and safety of intravenously administered tocilizumab was assessed in five randomized, double-blind, multicenter studies in patients greater than 18 years with active rheumatoid arthritis diagnosed according to American College of Rheumatology (ACR) criteria. Patients had at least 8 tender and 6 swollen joints at baseline. Tocilizumab was given intravenously every 4 weeks as monotherapy (Study I), in combination with methotrexate (MTX) (Studies II and III) or other disease-modifying anti-rheumatic drugs (DMARDs) (Study IV) in patients with an inadequate response to those drugs, or in combination with MTX in patients with an inadequate response to TNF antagonists (Study V).
Study I (NCT00109408) evaluated patients with moderate to severe active rheumatoid arthritis who had not been treated with MTX within 24 weeks prior to randomization, or who had not discontinued previous methotrexate treatment as a result of clinically important toxic effects or lack of response. In this study, 67% of patients were MTX-naïve, and over 40% of patients had rheumatoid arthritis less than 2 years. Patients received tocilizumab 8 mg per kg monotherapy or MTX alone (dose titrated over 8 weeks from 7.5 mg to a maximum of 20 mg weekly). The primary endpoint was the proportion of tocilizumab patients who achieved an ACR 20 response at Week 24.
Study II (NCT00106535) was a 104-week study with an optional 156-week extension phase that evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response to MTX. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). Upon completion of 52-weeks, patients received open-label treatment with tocilizumab 8 mg per kg through 104 weeks or they had the option to continue their double-blind treatment if they maintained a greater than 70% improvement in swollen/tender joint count. Two pre-specified interim analyses at week 24 and week 52 were conducted. The primary endpoint at week 24 was the proportion of patients who achieved an ACR 20 response. At weeks 52 and 104, the primary endpoints were change from baseline in modified total Sharp-Genant score and the area under the curve (AUC) of the change from baseline in HAQ-DI score.
Study III (NCT00106548) evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response to MTX. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
Study IV (NCT00106574) evaluated patients who had an inadequate response to their existing therapy, including one or more DMARDs. Patients received tocilizumab 8 mg per kg or placebo every four weeks, in combination with the stable DMARDs. The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
Study V (NCT00106522) evaluated patients with moderate to severe active rheumatoid arthritis who had an inadequate clinical response or were intolerant to one or more TNF antagonist therapies. The TNF antagonist therapy was discontinued prior to randomization. Patients received tocilizumab 8 mg per kg, tocilizumab 4 mg per kg, or placebo every four weeks, in combination with MTX (10 to 25 mg weekly). The primary endpoint was the proportion of patients who achieved an ACR 20 response at week 24.
14.2 Giant Cell Arteritis Intravenous Administration (14.2 Giant Cell Arteritis - Intravenous Administration)
Intravenously administered tocilizumab in patients with GCA was assessed in WP41152 (NCT03923738), an open-label PK-PD and safety study to determine the appropriate intravenous dose of tocilizumab that achieved comparable PK-PD profiles to tocilizumab by another route of administration.
At enrollment, all patients (n=24) were in remission on tocilizumab IV. In Period 1, all patients received open-label tocilizumab IV 7 mg/kg every 4 weeks for 20 weeks. Patients who completed Period 1 and remained in remission (n=22) were eligible to enter Period 2, and received open-label tocilizumab IV 6 mg/kg every 4 weeks for 20 weeks.
The efficacy of intravenous tocilizumab 6 mg/kg in adult patients with GCA is based on pharmacokinetic exposure and extrapolation to the efficacy established for tocilizumab by another route of administration in patients with GCA.
7.1 Concomitant Drugs for Treatment of Adult Indications
In RA patients, population pharmacokinetic analyses did not detect any effect of methotrexate (MTX), non-steroidal anti-inflammatory drugs or corticosteroids on tocilizumab clearance. Concomitant administration of a single intravenous dose of 10 mg/kg tocilizumab with 10-25 mg MTX once weekly had no clinically significant effect on MTX exposure. Tocilizumab products have not been studied in combination with biological DMARDs such as TNF antagonists [see Dosage and Administration (2.2)].
In GCA patients, no effect of concomitant corticosteroid on tocilizumab exposure was observed.
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term animal studies have been performed to establish the carcinogenicity potential of tocilizumab products. Literature indicates that the IL-6 pathway can mediate anti-tumor responses by promoting increased immune cell surveillance of the tumor microenvironment. However, available published evidence also supports that IL-6 signaling through the IL-6 receptor may be involved in pathways that lead to tumorigenesis. The malignancy risk in humans from an antibody that disrupts signaling through the IL-6 receptor, such as tocilizumab, is presently unknown.
Fertility and reproductive performance were unaffected in male and female mice that received a murine analogue of tocilizumab administered by the intravenous route at a dose of 50 mg/kg every three days.
2.5 Recommended Dosage for Systemic Juvenile Idiopathic Arthritis
TOFIDENCE may be used as an intravenous infusion or in combination with methotrexate. Do not change a dose based solely on a single visit body weight measurement, as weight may fluctuate.
2.4 Recommended Dosage for Polyarticular Juvenile Idiopathic Arthritis
TOFIDENCE may be used as an intravenous infusion alone or in combination with methotrexate. Do not change dose based solely on a single visit body weight measurement, as weight may fluctuate.
14.4 Systemic Juvenile Idiopathic Arthritis—intravenous Administration (14.4 Systemic Juvenile Idiopathic Arthritis—Intravenous Administration)
The efficacy of tocilizumab for the treatment of active SJIA was assessed in WA18221 (NCT00642460), a 12-week randomized, double blind, placebo-controlled, parallel group, 2-arm study. Patients treated with or without MTX, were randomized (tocilizumab:placebo = 2:1) to one of two treatment groups: 75 patients received tocilizumab infusions every two weeks at either 8 mg per kg for patients at or above 30 kg or 12 mg per kg for patients less than 30 kg and 37 were randomized to receive placebo infusions every two weeks. Corticosteroid tapering could occur from week six for patients who achieved a JIA ACR 70 response. After 12 weeks or at the time of escape, due to disease worsening, patients were treated with tocilizumab in the open-label extension phase at weight appropriate dosing.
The primary endpoint was the proportion of patients with at least 30% improvement in JIA ACR core set (JIA ACR 30 response) at Week 12 and absence of fever (no temperature at or above 37.5°C in the preceding 7 days). JIA ACR (American College of Rheumatology) responses are defined as the percentage improvement (e.g., 30%, 50%, 70%) in 3 of any 6 core outcome variables compared to baseline, with worsening in no more than 1 of the remaining variables by 30% or more. Core outcome variables consist of physician global assessment, parent per patient global assessment, number of joints with active arthritis, number of joints with limitation of movement, erythrocyte sedimentation rate (ESR), and functional ability (childhood health assessment questionnaire-CHAQ).
Primary endpoint result and JIA ACR response rates at Week 12 are shown in Table 8.
| Tocilizumab N=75 |
Placebo N=37 |
|
|---|---|---|
| Primary Endpoint: JIA ACR 30 response + absence of fever | ||
| Responders | 85% | 24% |
|
Weighted difference (95% CI) |
62 (45, 78) |
- |
| JIA ACR Response Rates at Week 12 | ||
| JIA ACR 30 | ||
| Responders | 91% | 24% |
| Weighted difference The weighted difference is the difference between the tocilizumab and Placebo response rates, adjusted for the stratification factors (weight, disease duration, background oral corticosteroid dose and background methotrexate use).
(95% CI) CI: confidence interval of the weighted difference.
|
67 (51, 83) |
- |
| JIA ACR 50 | ||
| Responders | 85% | 11% |
| Weighted difference
(95% CI) |
74 (58, 90) |
- |
| JIA ACR 70 | ||
| Responders | 71% | 8% |
| Weighted difference
(95% CI) |
63 (46, 80) |
- |
The treatment effect of tocilizumab was consistent across all components of the JIA ACR response core variables. JIA ACR scores and absence of fever responses in the open label extension were consistent with the controlled portion of the study (data available through 44 weeks).
2.7 Preparation and Administration Instructions for Intravenous Infusion
TOFIDENCE for intravenous infusion should be diluted by a healthcare professional using aseptic technique as follows:
- Use a sterile needle and syringe to prepare TOFIDENCE.
- Patients less than 30 kg: use a 50 mL infusion bag or bottle of 0.9% Sodium Chloride Injection, USP, and then follow steps 1 and 2 below.
- Patients at or above 30 kg weight: use a 100 mL infusion bag or bottle, and then follow steps 1 and 2 below.
- Step 1. Withdraw a volume of 0.9% Sodium Chloride Injection, USP, equal to the volume of the TOFIDENCE injection required for the patient’s dose from the infusion bag or bottle [see Dosage and Administration (2.2, 2.4, 2.5)].
| For Intravenous Use: Volume of TOFIDENCE Injection per kg of Body Weight | ||
|---|---|---|
| Dosage | Indication | Volume of TOFIDENCE injection per kg of body weight |
| 4 mg/kg | Adult RA | 0.2 mL/kg |
| 6 mg/kg | Adult GCA | 0.3 mL/kg |
| 8 mg/kg | Adult RA
Adult COVID-19 SJIA and PJIA (greater than or equal to 30 kg of body weight) |
0.4 mL/kg |
| 10 mg/kg | PJIA (less than 30 kg of body weight) | 0.5 mL/kg |
| 12 mg/kg | SJIA (less than 30 kg of body weight) | 0.6 mL/kg |
- Step 2. Withdraw the amount of TOFIDENCE for intravenous infusion from the vial(s) and add slowly into the 0.9% Sodium Chloride Injection, USP infusion bag or bottle. To mix the solution, gently invert the bag to avoid foaming.
- The fully diluted TOFIDENCE solutions for infusion using 0.9% Sodium Chloride Injection, USP may be stored refrigerated at 36°F to 46°F (2°C to 8°C) for up to 24 hours or room temperature at 68°F to 77°F (20°C to 25°C) for up to 12 hours and should be protected from light.
- TOFIDENCE solutions do not contain preservatives; therefore, unused product remaining in the vials should not be used.
- Allow the fully diluted TOFIDENCE solution to reach room temperature prior to infusion.
- The infusion should be administered over 60 minutes, and must be administered with an infusion set. Do not administer as an intravenous push or bolus.
- TOFIDENCE should not be infused concomitantly in the same intravenous line with other drugs. No physical or biochemical compatibility studies have been conducted to evaluate the co-administration of TOFIDENCE with other drugs.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If particulates and discolorations are noted, the product should not be used.
- Fully diluted TOFIDENCE solutions are compatible with infusion bags and/or infusion sets with the following materials: polypropylene, polyethylene, polyolefin, polyvinyl chloride, polyethersulfone, polyurethane, nylon and stainless steel.
14.3 Polyarticular Juvenile Idiopathic Arthritis—intravenous Administration (14.3 Polyarticular Juvenile Idiopathic Arthritis—Intravenous Administration)
The efficacy of tocilizumab was assessed in a three-part study, WA19977 (NCT00988221), including an open-label extension in children 2 to 17 years of age with active polyarticular juvenile idiopathic arthritis (PJIA), who had an inadequate response to methotrexate or inability to tolerate methotrexate. Patients had at least 6 months of active disease (mean disease duration of 4.2 ± 3.7 years), with at least five joints with active arthritis (swollen or limitation of movement accompanied by pain and/or tenderness) and/or at least 3 active joints having limitation of motion (mean, 20 ± 14 active joints). The patients treated had subtypes of JIA that at disease onset included Rheumatoid Factor Positive or Negative Polyarticular JIA, or Extended Oligoarticular JIA. Treatment with a stable dose of methotrexate was permitted but was not required during the study. Concurrent use of disease modifying antirheumatic drugs (DMARDs), other than methotrexate, or other biologics (e.g., TNF antagonists or T cell costimulation modulator) were not permitted in the study.
Part I consisted of a 16-week active tocilizumab treatment lead-in period (n=188) followed by Part II, a 24-week randomized double-blind placebo-controlled withdrawal period, followed by Part III, a 64-week open-label period. Eligible patients weighing at or above 30 kg received tocilizumab at 8 mg/kg intravenously once every four weeks. Patients weighing less than 30 kg were randomized 1:1 to receive either tocilizumab 8 mg/kg or 10 mg/kg intravenously every four weeks. At the conclusion of the open-label Part I, 91% of patients taking background MTX in addition to tocilizumab and 83% of patients on tocilizumab monotherapy achieved an ACR 30 response at week 16 compared to baseline and entered the blinded withdrawal period (Part II) of the study. The proportions of patients with JIA ACR 50/70 responses in Part I were 84.0%, and 64%, respectively for patients taking background MTX in addition to tocilizumab and 80% and 55% respectively for patients on tocilizumab monotherapy.
In Part II, patients (ITT, n=163) were randomized to tocilizumab (same dose received in Part I) or placebo in a 1:1 ratio that was stratified by concurrent methotrexate use and concurrent corticosteroid use. Each patient continued in Part II of the study until Week 40 or until the patient satisfied JIA ACR 30 flare criteria (relative to Week 16) and qualified for escape.
The primary endpoint was the proportion of patients with a JIA ACR 30 flare at week 40 relative to week 16. JIA ACR 30 flare was defined as 3 or more of the 6 core outcome variables worsening by at least 30% with no more than 1 of the remaining variables improving by more than 30% relative to Week 16.
Tocilizumab treated patients experienced significantly fewer disease flares compared to placebo-treated patients (26% [21/82] versus 48% [39/81]; adjusted difference in proportions -21%, 95% CI: -35%, -8%).
During the withdrawal phase (Part II), more patients treated with tocilizumab showed JIA ACR 30/50/70 responses at Week 40 compared to patients withdrawn to placebo.
6.5 Clinical Trials Experience in Covid 19 Patients Treated With Intravenous Tocilizumab (tocilizumab Iv) (6.5 Clinical Trials Experience in COVID-19 Patients Treated with Intravenous Tocilizumab (Tocilizumab-IV))
The safety of tocilizumab in hospitalized COVID-19 patients was evaluated in a pooled safety population that includes patients enrolled in EMPACTA, COVACTA, AND REMDACTA. The analysis of adverse reactions included a total of 974 patients exposed to tocilizumab. Patients received a single, 60-minute infusion of intravenous tocilizumab 8 mg/kg (maximum dose of 800 mg). If clinical signs or symptoms worsened or did not improve, one additional dose of tocilizumab 8 mg/kg could be administered between 8- 24 hours after the initial dose.
Adverse reactions summarized in Table 3 occurred in at least 3% of tocilizumab -treated patients and more commonly than in patients on placebo in the pooled safety population.
| Adverse Reaction | Tocilizumab 8mg per kg |
Placebo |
|---|---|---|
| N = 974 (%) |
N = 483 (%) |
|
| Hepatic Transaminases increased | 10% | 8% |
| Constipation | 9% | 8% |
| Urinary tract infection | 5% | 4% |
| Hypertension | 4% | 1% |
| Hypokalaemia | 4% | 3% |
| Anxiety | 4% | 2% |
| Diarrhea | 4% | 2% |
| Insomnia | 4% | 3% |
| Nausea | 3% | 2% |
In the pooled safety population, the rates of infection/serious infection events were 30%/19% in patients receiving tocilizumab versus 32%/23% receiving placebo.
6.1 Clinical Trials Experience in Rheumatoid Arthritis Patients Treated With Intravenous Tocilizumab (tocilizumab Iv) (6.1 Clinical Trials Experience in Rheumatoid Arthritis Patients Treated with Intravenous Tocilizumab (Tocilizumab-IV))
The tocilizumab-IV data in rheumatoid arthritis (RA) includes 5 double-blind, controlled, multicenter studies. In these studies, patients received doses of tocilizumab-IV 8 mg per kg monotherapy (288 patients), tocilizumab-IV 8 mg per kg in combination with DMARDs (including methotrexate) (1582 patients), or tocilizumab-IV 4 mg per kg in combination with methotrexate (774 patients).
The all exposure population includes all patients in registration studies who received at least one dose of tocilizumab-IV. Of the 4009 patients in this population, 3577 received treatment for at least 6 months, 3309 for at least one year; 2954 received treatment for at least 2 years and 2189 for 3 years.
All patients in these studies had moderately to severely active rheumatoid arthritis. The study population had a mean age of 52 years, 82% were female and 74% were Caucasian.
The most common serious adverse reactions were serious infections [see Warnings and Precautions (5.1)]. The most commonly reported adverse reactions in controlled studies up to 24 weeks (occurring in at least 5% of patients treated with tocilizumab-IV monotherapy or in combination with DMARDs) were upper respiratory tract infections, nasopharyngitis, headache, hypertension and increased ALT.
The proportion of patients who discontinued treatment due to any adverse reactions during the double-blind, placebo-controlled studies was 5% for patients taking tocilizumab-IV and 3% for placebo-treated patients. The most common adverse reactions that required discontinuation of tocilizumab-IV were increased hepatic transaminase values (per protocol requirement) and serious infections.
6.2 Clinical Trials Experience in Giant Cell Arteritis Patients Treated With Intravenous Tocilizumab (tocilizumab Iv) (6.2 Clinical Trials Experience in Giant Cell Arteritis Patients Treated with Intravenous Tocilizumab (Tocilizumab-IV))
The safety of tocilizumab-IV was studied in an open label PK-PD and safety study in 24 patients with GCA who were in remission on tocilizumab-IV at time of enrollment. Patients received tocilizumab 7 mg/kg every 4 weeks for 20 weeks, followed by 6 mg/kg every 4 weeks for 20 weeks. The total patient years exposure to treatment was 17.5 years.
The safety of tocilizumab by another route of administration has been studied in one Phase III study with 251 GCA patients. The total patient years duration was 138.5 patient years during the 12-month double blind, placebo-controlled phase of the study. The overall safety profile observed was generally consistent with the known safety profile of tocilizumab. There was an overall higher incidence of infections in GCA patients relative to RA patients.
The overall safety profile observed for tocilizumab administered intravenously in GCA patients was consistent with the known safety profile of tocilizumab.
6.4 Clinical Trials Experience in Systemic Juvenile Idiopathic Arthritis Patients Treated With Intravenous Tocilizumab (tocilizumab Iv) (6.4 Clinical Trials Experience in Systemic Juvenile Idiopathic Arthritis Patients Treated with Intravenous Tocilizumab (Tocilizumab-IV))
The data described below reflect exposure to tocilizumab-IV in one randomized, double-blind, placebo-controlled trial of 112 pediatric patients with SJIA 2 to 17 years of age who had an inadequate clinical response to nonsteroidal anti-inflammatory drugs (NSAIDs) or corticosteroids due to toxicity or lack of efficacy. At baseline, approximately half of the patients were taking 0.3 mg/kg/day corticosteroids or more, and almost 70% were taking methotrexate. The trial included a 12 week controlled phase followed by an open-label extension. In the 12 week double-blind, controlled portion of the clinical study 75 patients received treatment with tocilizumab-IV (8 or 12 mg per kg based upon body weight). After 12 weeks or at the time of escape, due to disease worsening, patients were treated with tocilizumab-IV in the open-label extension phase.
The most common adverse events (at least 5%) seen in tocilizumab-IV treated patients in the 12 week controlled portion of the study were: upper respiratory tract infection, headache, nasopharyngitis and diarrhea.
6.3 Clinical Trials Experience in Polyarticular Juvenile Idiopathic Arthritis Patients Treated With Intravenous Tocilizumab (tocilizumab Iv) (6.3 Clinical Trials Experience in Polyarticular Juvenile Idiopathic Arthritis Patients Treated with Intravenous Tocilizumab (Tocilizumab-IV))
The safety of tocilizumab-IV was studied in 188 pediatric patients 2 to 17 years of age with PJIA who had an inadequate clinical response or were intolerant to methotrexate. The total patient exposure in the tocilizumab-IV all exposure population (defined as patients who received at least one dose of tocilizumab-IV) was 184.4 patient years. At baseline, approximately half of the patients were taking oral corticosteroids and almost 80% were taking methotrexate. In general, the types of adverse drug reactions in patients with PJIA were consistent with those seen in RA and SJIA patients [see Adverse Reactions (6.1 and 6.4)].
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:49:44.392902 · Updated: 2026-03-14T22:34:21.968394