Ellence
0a03c798-a652-4895-b29c-3b521a89ba42
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
ELLENCE is indicated as a component of adjuvant therapy in patients with evidence of axillary node tumor involvement following resection of primary breast cancer [see Clinical Studies (14.1) ] .
Dosage and Administration
• The recommended starting dose of ELLENCE is 100 to 120 mg/m 2 . Dosage reductions are possible when given in certain combinations ( 2.2 ). • Administer intravenously in repeated 3- to 4-week cycles, either total dose on Day 1 of each cycle or divided equally and given on Days 1 and 8 of each cycle ( 2.2 ). • Consider use of antiemetics when given in conjunction with other emetigenic drugs ( 2.1 ). • Patients administered the 120 mg/m 2 regimen of ELLENCE should receive prophylactic antibiotic therapy ( 2.1 ). • Adjust dosage after the first treatment cycle based on hematologic and nonhematologic toxicities ( 2.3 ). • Reduce dose in patients with hepatic impairment ( 2.3 , 8.6 ). • Consider lower doses in patients with severe renal impairment ( 2.3 , 8.7 ).
Contraindications
ELLENCE is contraindicated in patients with: • Severe myocardial insufficiency [see Warnings and Precautions (5.1) ] • Recent myocardial infarction or severe arrhythmias, or previous treatment with maximum cumulative dose of anthracyclines [see Warnings and Precautions (5.1) ] • Severe persistent drug-induced myelosuppression [see Warnings and Precautions (5.4) ] • Severe hepatic impairment (defined as Child-Pugh Class C or serum bilirubin level greater than 5 mg/dL) [see Warnings and Precautions (5.5) ] • Severe hypersensitivity to ELLENCE, other anthracyclines, or anthracenediones [see Adverse Reactions (6.1) ]
Warnings and Precautions
• Use in Patients with Hepatic Impairment: Monitor serum total bilirubin and AST levels before and during treatment with ELLENCE. In patients with elevated serum AST or serum total bilirubin, dosage reductions or discontinuation may be required ( 2.3 , 5.5 ). • Tumor Lysis Syndrome: Evaluate blood uric acid levels, potassium, calcium, phosphate, and creatinine after initial treatment. Consider hydration, urine alkalinization, and prophylaxis with allopurinol to minimize potential complications of hyperuricemia and tumor lysis syndrome ( 5.7 ). • Thrombophlebitis and Thromboembolic Events: Thrombophlebitis and thromboembolic events, including pulmonary embolism (in some cases fatal) have been reported with the use of ELLENCE. Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein ( 5.9 ). • Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including ELLENCE, may result in serious or fatal infections ( 5.7 ). • Potentiation of Radiation Toxicity and Radiation Recall: Administration of ELLENCE after previous radiation therapy may induce an inflammatory recall reaction at the site of the irradiation ( 5.10 ). • Embryo-Fetal Toxicity: ELLENCE can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception ( 5.11 , 8.1 , 8.3 ).
Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling: • Cardiac Toxicity [see Warnings and Precautions (5.1) ] • Secondary Malignancies [see Warnings and Precautions (5.2) ] • Extravasation and Tissue Necrosis [see Warnings and Precautions (5.3) ] • Severe Myelosuppression [see Warnings and Precautions (5.4) ] • Tumor-Lysis Syndrome [see Warnings and Precautions (5.7) ] • Thrombophlebitis and Thromboembolic Events [see Warnings and Precautions (5.9) ] • Potentiation of Radiation Toxicity and Radiation Recall [see Warnings and Precautions (5.10) ]
Drug Interactions
• Avoid using cardiotoxic agents in combination with ELLENCE ( 7.1 ). • Discontinue cimetidine during treatment with ELLENCE ( 7.2 ).
How Supplied
ELLENCE is available in polypropylene single-dose CYTOSAFE ® vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths: Unit of Sale Strength NDC 0009-5091-01 Carton of 1 Single-dose Vial 50 mg/25 mL (2 mg/mL) NDC 0009-5093-01 Carton of 1 Single-dose Vial 200 mg/100 mL (2 mg/mL) Discard unused portion. ELLENCE is available in ONCO-TAIN ® glass vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths: Unit of Sale Strength NDC 0009-5091-25 Carton of 1 Single-dose Vial 50 mg/25 mL (2 mg/mL) NDC 0009-5093-10 Carton of 1 Single-dose Vial 200 mg/100 mL (2 mg/mL) ONCO-TAIN ® is the vial external protection system. Discard unused portion.
Storage and Handling
ELLENCE is available in polypropylene single-dose CYTOSAFE ® vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths: Unit of Sale Strength NDC 0009-5091-01 Carton of 1 Single-dose Vial 50 mg/25 mL (2 mg/mL) NDC 0009-5093-01 Carton of 1 Single-dose Vial 200 mg/100 mL (2 mg/mL) Discard unused portion. ELLENCE is available in ONCO-TAIN ® glass vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths: Unit of Sale Strength NDC 0009-5091-25 Carton of 1 Single-dose Vial 50 mg/25 mL (2 mg/mL) NDC 0009-5093-10 Carton of 1 Single-dose Vial 200 mg/100 mL (2 mg/mL) ONCO-TAIN ® is the vial external protection system. Discard unused portion.
Description
• Cardiac Toxicity: Myocardial damage, including acute left ventricular failure, can occur with ELLENCE. The risk of cardiomyopathy is proportional to the cumulative exposure with incidence rates from 0.9% at a cumulative dose of 550 mg/m 2 , 1.6% at 700 mg/m 2 , and 3.3% at 900 mg/m 2 . The risk of cardiomyopathy is further increased with concomitant cardiotoxic therapy. Assess left ventricular ejection fraction (LVEF) before and regularly during and after treatment with ELLENCE [see Warnings and Precautions (5.1) ] . • Secondary Malignancies: Secondary acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) occur at a higher incidence in patients treated with anthracyclines, including ELLENCE [see Warnings and Precautions (5.2) ] . • Extravasation and Tissue Necrosis: Extravasation of ELLENCE can result in severe local tissue injury and necrosis requiring wide excision of the affected area and skin grafting. Immediately terminate the drug and apply ice to the affected area [see Warnings and Precautions (5.3) ] . • Severe myelosuppression resulting in serious infection, septic shock, requirement for transfusions, hospitalization, and death may occur [see Warnings and Precautions (5.4) ] .
Medication Information
Warnings and Precautions
• Use in Patients with Hepatic Impairment: Monitor serum total bilirubin and AST levels before and during treatment with ELLENCE. In patients with elevated serum AST or serum total bilirubin, dosage reductions or discontinuation may be required ( 2.3 , 5.5 ). • Tumor Lysis Syndrome: Evaluate blood uric acid levels, potassium, calcium, phosphate, and creatinine after initial treatment. Consider hydration, urine alkalinization, and prophylaxis with allopurinol to minimize potential complications of hyperuricemia and tumor lysis syndrome ( 5.7 ). • Thrombophlebitis and Thromboembolic Events: Thrombophlebitis and thromboembolic events, including pulmonary embolism (in some cases fatal) have been reported with the use of ELLENCE. Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein ( 5.9 ). • Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including ELLENCE, may result in serious or fatal infections ( 5.7 ). • Potentiation of Radiation Toxicity and Radiation Recall: Administration of ELLENCE after previous radiation therapy may induce an inflammatory recall reaction at the site of the irradiation ( 5.10 ). • Embryo-Fetal Toxicity: ELLENCE can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception ( 5.11 , 8.1 , 8.3 ).
Indications and Usage
ELLENCE is indicated as a component of adjuvant therapy in patients with evidence of axillary node tumor involvement following resection of primary breast cancer [see Clinical Studies (14.1) ] .
Dosage and Administration
• The recommended starting dose of ELLENCE is 100 to 120 mg/m 2 . Dosage reductions are possible when given in certain combinations ( 2.2 ). • Administer intravenously in repeated 3- to 4-week cycles, either total dose on Day 1 of each cycle or divided equally and given on Days 1 and 8 of each cycle ( 2.2 ). • Consider use of antiemetics when given in conjunction with other emetigenic drugs ( 2.1 ). • Patients administered the 120 mg/m 2 regimen of ELLENCE should receive prophylactic antibiotic therapy ( 2.1 ). • Adjust dosage after the first treatment cycle based on hematologic and nonhematologic toxicities ( 2.3 ). • Reduce dose in patients with hepatic impairment ( 2.3 , 8.6 ). • Consider lower doses in patients with severe renal impairment ( 2.3 , 8.7 ).
Contraindications
ELLENCE is contraindicated in patients with: • Severe myocardial insufficiency [see Warnings and Precautions (5.1) ] • Recent myocardial infarction or severe arrhythmias, or previous treatment with maximum cumulative dose of anthracyclines [see Warnings and Precautions (5.1) ] • Severe persistent drug-induced myelosuppression [see Warnings and Precautions (5.4) ] • Severe hepatic impairment (defined as Child-Pugh Class C or serum bilirubin level greater than 5 mg/dL) [see Warnings and Precautions (5.5) ] • Severe hypersensitivity to ELLENCE, other anthracyclines, or anthracenediones [see Adverse Reactions (6.1) ]
Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling: • Cardiac Toxicity [see Warnings and Precautions (5.1) ] • Secondary Malignancies [see Warnings and Precautions (5.2) ] • Extravasation and Tissue Necrosis [see Warnings and Precautions (5.3) ] • Severe Myelosuppression [see Warnings and Precautions (5.4) ] • Tumor-Lysis Syndrome [see Warnings and Precautions (5.7) ] • Thrombophlebitis and Thromboembolic Events [see Warnings and Precautions (5.9) ] • Potentiation of Radiation Toxicity and Radiation Recall [see Warnings and Precautions (5.10) ]
Drug Interactions
• Avoid using cardiotoxic agents in combination with ELLENCE ( 7.1 ). • Discontinue cimetidine during treatment with ELLENCE ( 7.2 ).
Storage and Handling
ELLENCE is available in polypropylene single-dose CYTOSAFE ® vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths: Unit of Sale Strength NDC 0009-5091-01 Carton of 1 Single-dose Vial 50 mg/25 mL (2 mg/mL) NDC 0009-5093-01 Carton of 1 Single-dose Vial 200 mg/100 mL (2 mg/mL) Discard unused portion. ELLENCE is available in ONCO-TAIN ® glass vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths: Unit of Sale Strength NDC 0009-5091-25 Carton of 1 Single-dose Vial 50 mg/25 mL (2 mg/mL) NDC 0009-5093-10 Carton of 1 Single-dose Vial 200 mg/100 mL (2 mg/mL) ONCO-TAIN ® is the vial external protection system. Discard unused portion.
How Supplied
ELLENCE is available in polypropylene single-dose CYTOSAFE ® vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths: Unit of Sale Strength NDC 0009-5091-01 Carton of 1 Single-dose Vial 50 mg/25 mL (2 mg/mL) NDC 0009-5093-01 Carton of 1 Single-dose Vial 200 mg/100 mL (2 mg/mL) Discard unused portion. ELLENCE is available in ONCO-TAIN ® glass vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths: Unit of Sale Strength NDC 0009-5091-25 Carton of 1 Single-dose Vial 50 mg/25 mL (2 mg/mL) NDC 0009-5093-10 Carton of 1 Single-dose Vial 200 mg/100 mL (2 mg/mL) ONCO-TAIN ® is the vial external protection system. Discard unused portion.
Description
• Cardiac Toxicity: Myocardial damage, including acute left ventricular failure, can occur with ELLENCE. The risk of cardiomyopathy is proportional to the cumulative exposure with incidence rates from 0.9% at a cumulative dose of 550 mg/m 2 , 1.6% at 700 mg/m 2 , and 3.3% at 900 mg/m 2 . The risk of cardiomyopathy is further increased with concomitant cardiotoxic therapy. Assess left ventricular ejection fraction (LVEF) before and regularly during and after treatment with ELLENCE [see Warnings and Precautions (5.1) ] . • Secondary Malignancies: Secondary acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) occur at a higher incidence in patients treated with anthracyclines, including ELLENCE [see Warnings and Precautions (5.2) ] . • Extravasation and Tissue Necrosis: Extravasation of ELLENCE can result in severe local tissue injury and necrosis requiring wide excision of the affected area and skin grafting. Immediately terminate the drug and apply ice to the affected area [see Warnings and Precautions (5.3) ] . • Severe myelosuppression resulting in serious infection, septic shock, requirement for transfusions, hospitalization, and death may occur [see Warnings and Precautions (5.4) ] .
Section 42229-5
Cardiac Toxicity
Discontinue ELLENCE in patients who develop signs or symptoms of cardiomyopathy [see Warnings and Precautions (5.1)].
Section 44425-7
Store refrigerated between 2°C and 8°C (36°F and 46°F). Do not freeze. Protect from light.
Storage of the solution for injection at refrigerated conditions can result in the formation of a gelled product. This gelled product will return to a slightly viscous to mobile solution after 2 to a maximum of 4 hours equilibration at controlled room temperature (15–25°C). Solution for injection should be used within 24 hours after removal from refrigeration.
ELLENCE is a hazardous drug. Follow applicable special handling and disposal procedures1 [see References (15)].
10 Overdosage
There is no known antidote for overdoses of ELLENCE. A 36-year-old man with non-Hodgkin's lymphoma received a daily 95 mg/m2 dose of ELLENCE for 5 consecutive days. Five days later, he developed bone marrow aplasia, grade 4 mucositis, and gastrointestinal bleeding. No signs of acute cardiac toxicity were observed. He was treated with antibiotics, colony-stimulating factors, and antifungal agents, and recovered completely. A 63-year-old woman with breast cancer and liver metastasis received a single 320 mg/m2 dose of ELLENCE. She was hospitalized with hyperthermia and developed multiple organ failure (respiratory and renal), with lactic acidosis, increased lactate dehydrogenase, and anuria. Death occurred within 24 hours after administration of ELLENCE. Additional instances of administration of doses higher than recommended have been reported at doses ranging from 150 to 250 mg/m2. The observed adverse events in these patients were qualitatively similar to known toxicities of epirubicin. Most of the patients recovered with appropriate supportive care.
If an overdose occurs, provide supportive treatment (including antibiotic therapy, blood and platelet transfusions, colony-stimulating factors, and intensive care as needed) until the recovery of toxicities. Delayed CHF has been observed months after anthracycline administration. Observe patients carefully over time for signs of CHF and provided with appropriate supportive therapy.
15 References
-
1."Hazardous Drugs". OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
11 Description
ELLENCE (epirubicin hydrochloride injection) is an anthracycline topoisomerase inhibitor for intravenous administration. ELLENCE is supplied as a sterile, clear, red solution and is available in polypropylene vials containing 50 and 200 mg of epirubicin hydrochloride as a preservative-free, ready-to-use solution. Each milliliter of solution contains 2 mg of epirubicin hydrochloride. Inactive ingredients include 9 mg sodium chloride, USP, and water for injection, USP. The pH of the solution has been adjusted to 3.0 with hydrochloric acid and/or sodium hydroxide, NF.
Epirubicin hydrochloride is the 4-epimer of doxorubicin and is a semi-synthetic derivative of daunorubicin. The chemical name is (8S- cis )-10-[(3-amino-2,3,6-trideoxy-α-L- arabino -hexopyranosyl)oxy]-7,8,9,10-tetrahydro6,8,11-trihydroxy-8-(hydroxyacetyl)-1-methoxy-5,12-naphthacenedione hydrochloride. The active ingredient is a red-orange hygroscopic powder, with the empirical formula C27 H29 NO11 HCl and a molecular weight of 579.95. The structural formula is as follows:
7.2 Cimetidine
Cimetidine increases the exposure to epirubicin [see Clinical Pharmacology (12.3)]. Discontinue cimetidine during treatment with ELLENCE.
8.4 Pediatric Use
Safety and effectiveness of ELLENCE have not been established in pediatric patients. Pediatric patients may be at greater risk for anthracycline-induced acute manifestations of cardiotoxicity or late cardiovascular dysfunction. The pharmacokinetics of epirubicin in pediatric patients have not been evaluated.
8.5 Geriatric Use
Clinical experience in patients who were 65 years of age and older who received ELLENCE chemotherapy regimens for primary breast cancer showed no overall differences in safety and effectiveness compared with younger patients.
In elderly female patients, closely monitor for increased toxicity due to the risk of decreased clearance of epirubicin [see Clinical Pharmacology (12.3)].
4 Contraindications
ELLENCE is contraindicated in patients with:
-
•Severe myocardial insufficiency [see Warnings and Precautions (5.1)]
-
•Recent myocardial infarction or severe arrhythmias, or previous treatment with maximum cumulative dose of anthracyclines [see Warnings and Precautions (5.1)]
-
•Severe persistent drug-induced myelosuppression [see Warnings and Precautions (5.4)]
-
•Severe hepatic impairment (defined as Child-Pugh Class C or serum bilirubin level greater than 5 mg/dL) [see Warnings and Precautions (5.5)]
-
•Severe hypersensitivity to ELLENCE, other anthracyclines, or anthracenediones [see Adverse Reactions (6.1)]
6 Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling:
-
•Cardiac Toxicity [see Warnings and Precautions (5.1)]
-
•Secondary Malignancies [see Warnings and Precautions (5.2)]
-
•Extravasation and Tissue Necrosis [see Warnings and Precautions (5.3)]
-
•Severe Myelosuppression [see Warnings and Precautions (5.4)]
-
•Tumor-Lysis Syndrome [see Warnings and Precautions (5.7)]
-
•Thrombophlebitis and Thromboembolic Events [see Warnings and Precautions (5.9)]
-
•Potentiation of Radiation Toxicity and Radiation Recall [see Warnings and Precautions (5.10)]
7 Drug Interactions
2.2 Recommended Dose
The recommended dose of ELLENCE is 100 to 120 mg/m2 administered as an intravenous bolus [see Dosage and Administration (2.4)].
The following regimens are recommended:
|
CEF-120: |
Cyclophosphamide |
75 mg/m2 oral on Days 1 to 14 |
|
ELLENCE |
60 mg/m2 intravenously on Days 1 and 8 |
|
|
5-Fluorouracil |
500 mg/m2 intravenously on Days 1 and 8 |
|
|
Repeat every 28 days for 6 cycles |
||
|
FEC-100: |
5-Fluorouracil |
500 mg/m2 intravenously on Day 1 |
|
ELLENCE |
100 mg/m2 intravenously on Day 1 |
|
|
Cyclophosphamide |
500 mg/m2 intravenously on Day 1 |
|
|
Repeat every 21 days for 6 cycles |
Administer ELLENCE in repeated 3- to 4-week cycles. The total dose of ELLENCE may be given on Day 1 of each cycle or divided equally and given on Days 1 and 8 of each cycle.
5.1 Cardiac Toxicity
ELLENCE and other anthracycline drugs can result in either early (or acute) or late (delayed) cardiac toxicity.
The principal manifestations of early cardiac toxicity are sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes. However, tachycardia (including premature ventricular contractions and ventricular tachycardia), bradycardia, as well as atrioventricular and bundle-branch block have been reported. Early cardiac toxicity does not usually predict the subsequent occurrence of delayed cardiotoxicity and generally should not be considered a reason for suspending treatment with ELLENCE.
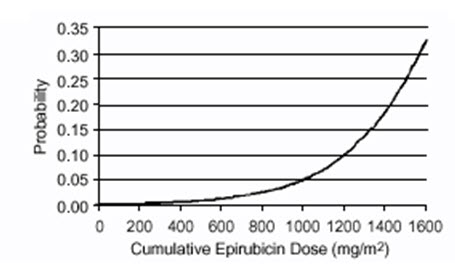
Delayed cardiac toxicity is manifested by reduced left ventricular ejection fraction (LVEF) and/or signs and symptoms of congestive heart failure (CHF). If it occurs, late cardiotoxicity usually develops late during therapy with ELLENCE or within 2 to 3 months after completion of treatment, but there are reports of it occurring several months to years after treatment termination. In a retrospective survey, including 9144 patients, mostly with solid tumors in advanced stages, the probability of developing CHF increased with increasing cumulative doses of ELLENCE (Figure 1). In another retrospective survey of 469 ELLENCE-treated patients with metastatic or early breast cancer, the reported risk of CHF was comparable to that observed in the larger study of over 9000 patients.
Given the risk of cardiac toxicity, cumulative doses of 900 mg/m2 ELLENCE should generally be avoided.
Figure 1. Risk of CHF in 9144 Patients Treated with ELLENCE
Prior history of cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, and concomitant use of other cardiotoxic drugs, increase the risk of developing late cardiac toxicity. Avoid administration of ELLENCE in combination with other cardiotoxic drugs. Although not formally tested, it is probable that the toxicity of ELLENCE and other anthracyclines or anthracenediones is additive. Cardiac toxicity with ELLENCE may occur at lower cumulative doses whether or not cardiac risk factors are present. Patients receiving ELLENCE after stopping treatment with other cardiotoxic drugs, especially those with long half-lives such as trastuzumab, may be at increased risk of developing cardiotoxicity [see Dosage and Administration (2) and Drug Interaction (7.1)].
Perform a baseline ECG and evaluation of LVEF prior to initiating treatment with ELLENCE. Monitor LVEF during the course of treatment and consider discontinuation of ELLENCE if LVEF decrease and/or signs or symptoms of CHF develop. Closely monitor patients with other risk factors for cardiac toxicity, particularly prior administration of anthracycline or anthracenedione.
8.7 Renal Impairment
No significant alterations in the pharmacokinetics of epirubicin or its major metabolite, epirubicinol, have been observed in patients with serum creatinine <5 mg/dL. Consider lower doses in patients with severe renal impairment (serum creatinine >5 mg/dL), as a reduction in plasma clearance was reported in these patients [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)]. Patients on dialysis have not been studied.
12.3 Pharmacokinetics
Epirubicin pharmacokinetics are linear over the dose range of 60 to 150 mg/m2 and plasma clearance is not affected by the duration of infusion or administration schedule. Pharmacokinetic parameters for epirubicin following 6- to 10-minute, single-dose intravenous infusions of ELLENCE at doses of 60 to 150 mg/m2 in patients with solid tumors are shown in Table 4. The plasma concentration declined in a triphasic manner with mean half-lives for the alpha, beta, and gamma phases of about 3 minutes, 2.5 hours, and 33 hours, respectively.
|
Dose
N=6 patients per dose level
(mg/m2) |
Cmax
Plasma concentration at the end of 6 to 10 minutes infusion
(µg/mL) |
AUC
Area under the plasma concentration curve
(µg∙h/mL) |
t1/2
Half-life of terminal phase
(hours) |
CL
Plasma clearance
(L/hour) |
Vss
Steady state volume of distribution
(L/kg) |
|---|---|---|---|---|---|
|
60 |
5.7 ± 1.6 |
1.6 ± 0.2 |
35.3 ± 9 |
65 ± 8 |
21 ± 2 |
|
75 |
5.3 ± 1.5 |
1.7 ± 0.3 |
32.1 ± 5 |
83 ± 14 |
27 ± 11 |
|
120 |
9.0 ± 3.5 |
3.4 ± 0.7 |
33.7 ± 4 |
65 ± 13 |
23 ± 7 |
|
150 |
9.3 ± 2.9 |
4.2 ± 0.8 |
31.1 ± 6 |
69 ± 13 |
21 ± 7 |
7.4 Radiation Therapy
There are few data regarding the coadministration of radiation therapy and ELLENCE. In adjuvant trials of ELLENCE-containing CEF-120 or FEC-100 chemotherapies, breast irradiation was delayed until after chemotherapy was completed. This practice resulted in no apparent increase in local breast cancer recurrence relative to published accounts in the literature. A small number of patients received ELLENCE-based chemotherapy concomitantly with radiation therapy but had chemotherapy interrupted in order to avoid potential overlapping toxicities. It is likely that use of ELLENCE with radiotherapy may sensitize tissues to the cytotoxic actions of irradiation. Administration of ELLENCE after previous radiation therapy may induce an inflammatory recall reaction at the site of the irradiation.
2.3 Dose Modifications
ELLENCE dosage adjustments for hematologic and non-hematologic toxicities within a cycle of treatment, is based on nadir platelet counts <50,000/mm3, absolute neutrophil counts (ANC) <250/mm3, neutropenic fever, or Grades 3/4 nonhematologic toxicity. Reduce ELLENCE Day 1 dose in subsequent cycles to 75% of the Day 1 dose given in the current cycle. Delay Day 1 chemotherapy in subsequent courses of treatment until platelet counts are ≥100,000/mm3, ANC ≥1500/mm3, and nonhematologic toxicities have recovered to ≤ Grade 1.
7.1 Cardiotoxic Agents
Closely monitor cardiac function when ELLENCE is used in combination with other cardiotoxic agents. Patients receiving ELLENCE after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, may be at an increased risk of developing cardiotoxicity [see Dosage and Administration (2) and Warnings and Precautions (5.1)]. Trastuzumab may persist in the circulation for up to 7 months. Therefore, avoid anthracycline-based therapy for up to 7 months after stopping trastuzumab when possible. Monitor the patient's cardiac function closely if anthracyclines are used before this time.
Concomitant use of ELLENCE with other cardioactive compounds that could cause heart failure (e.g., calcium channel blockers), requires close monitoring of cardiac function throughout treatment.
8.6 Hepatic Impairment
Epirubicin is eliminated by both hepatic metabolism and biliary excretion and clearance is reduced in patients with hepatic dysfunction. Do not treat patients with severe hepatic impairment with ELLENCE [see Contraindications (4)]. Reduce the starting dose for patients with less severe hepatic impairment [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
1 Indications and Usage
ELLENCE is indicated as a component of adjuvant therapy in patients with evidence of axillary node tumor involvement following resection of primary breast cancer [see Clinical Studies (14.1)].
12.1 Mechanism of Action
Epirubicin is an anthracycline cytotoxic agent. Although it is known that anthracyclines can interfere with a number of biochemical and biological functions within eukaryotic cells, the precise mechanisms of epirubicin's cytotoxic and/or antiproliferative properties have not been completely elucidated.
Epirubicin forms a complex with DNA by intercalation of its planar rings between nucleotide base pairs, with consequent inhibition of nucleic acid (DNA and RNA) and protein synthesis.
Such intercalation triggers DNA cleavage by topoisomerase II, resulting in cytocidal activity. Epirubicin also inhibits DNA helicase activity, preventing the enzymatic separation of double-stranded DNA and interfering with replication and transcription. Epirubicin is also involved in oxidation/reduction reactions by generating cytotoxic free radicals. The antiproliferative and cytotoxic activity of epirubicin is thought to result from these or other possible mechanisms.
Epirubicin is cytotoxic in vitro to a variety of established murine and human cell lines and primary cultures of human tumors. It is also active in vivo against a variety of murine tumors and human xenografts in athymic mice, including breast tumors.
5.7 Tumor Lysis Syndrome
ELLENCE can induce tumor lysis syndrome in patients with rapidly growing tumors. Evaluate blood uric acid levels, potassium, calcium, phosphate, and creatinine after initial treatment. Consider hydration, urine alkalinization, and prophylaxis with allopurinol to minimize hyperuricemia and potential complications of tumor lysis syndrome.
7.3 Other Cytotoxic Drugs
ELLENCE used in combination with other cytotoxic drugs may show on-treatment additive toxicity, especially hematologic and gastrointestinal effects.
5 Warnings and Precautions
-
•Use in Patients with Hepatic Impairment: Monitor serum total bilirubin and AST levels before and during treatment with ELLENCE. In patients with elevated serum AST or serum total bilirubin, dosage reductions or discontinuation may be required (2.3, 5.5).
-
•Tumor Lysis Syndrome: Evaluate blood uric acid levels, potassium, calcium, phosphate, and creatinine after initial treatment. Consider hydration, urine alkalinization, and prophylaxis with allopurinol to minimize potential complications of hyperuricemia and tumor lysis syndrome (5.7).
-
•Thrombophlebitis and Thromboembolic Events: Thrombophlebitis and thromboembolic events, including pulmonary embolism (in some cases fatal) have been reported with the use of ELLENCE. Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein (5.9).
-
•Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including ELLENCE, may result in serious or fatal infections (5.7).
-
•Potentiation of Radiation Toxicity and Radiation Recall: Administration of ELLENCE after previous radiation therapy may induce an inflammatory recall reaction at the site of the irradiation (5.10).
-
•Embryo-Fetal Toxicity: ELLENCE can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception (5.11, 8.1, 8.3).
5.11 Embryo Fetal Toxicity
Based on findings from animals and its mechanism of action, ELLENCE can cause fetal harm when administered to a pregnant woman; avoid the use of ELLENCE during the 1st trimester. Available human data do not establish the presence or absence of major birth defects and miscarriage related to the use of epirubicin during the 2nd and 3rd trimesters. In animal reproduction studies, epirubicin was embryo-fetal lethal and caused structural abnormalities in rats and rabbits at doses less than the maximum recommended human dose on a body surface area basis. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise female patients of reproductive potential to use effective contraception during treatment and for 6 months after the last dose of ELLENCE. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ELLENCE. Advise male patients with pregnant partners to use condoms during treatment and for at least 7 days after the last dose of ELLENCE [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1), and Nonclinical Toxicology (13.1)].
5.2 Secondary Malignancies
The risk of developing secondary acute myelogenous leukemia and myelodysplastic syndrome (MDS), is increased following treatment with ELLENCE and other anthracyclines. Cumulative risk of secondary acute myelogenous leukemia or myelodysplastic syndrome (AML/MDS) of about 0.27% at 3 years, 0.46% at 5 years, and 0.55% at 8 years. These leukemias generally occur within 1 to 3 years of treatment [see Adverse Reactions (6.1)].
2 Dosage and Administration
-
•The recommended starting dose of ELLENCE is 100 to 120 mg/m2. Dosage reductions are possible when given in certain combinations (2.2).
-
•Administer intravenously in repeated 3- to 4-week cycles, either total dose on Day 1 of each cycle or divided equally and given on Days 1 and 8 of each cycle (2.2).
-
•Consider use of antiemetics when given in conjunction with other emetigenic drugs (2.1).
-
•Patients administered the 120 mg/m2 regimen of ELLENCE should receive prophylactic antibiotic therapy (2.1).
-
•Adjust dosage after the first treatment cycle based on hematologic and nonhematologic toxicities (2.3).
-
•Reduce dose in patients with hepatic impairment (2.3, 8.6).
-
•Consider lower doses in patients with severe renal impairment (2.3, 8.7).
5.4 Severe Myelosuppression
ELLENCE can cause severe myelosuppression [see Adverse Reactions (6.1) . Obtain complete blood counts prior to each treatment and carefully monitor patients during treatment for possible clinical complications due to myelosuppression. Delay the next dose of ELLENCE if severe myelosuppression has not improved. Consider dose reduction for patients with prolonged myelosuppression based on the severity of reaction [see Dosage and Administration (2.3)].
3 Dosage Forms and Strengths
Injection: 50 mg/25 mL (2 mg/mL), 200 mg/100 mL (2 mg/mL) clear red solution in a single-dose vial.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ELLENCE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Infections and infestations: sepsis, pneumonia
Immune system disorders: anaphylaxis
Metabolism and nutrition disorders: dehydration, hyperuricemia
Vascular disorders: shock, hemorrhage, embolism arterial, thrombophlebitis, phlebitis
Respiratory, thoracic and mediastinal disorders: pulmonary embolism
Gastrointestinal disorders: erosions, ulcerations, pain or burning sensation, bleeding, hyperpigmentation of the oral mucosa
Skin and subcutaneous tissue disorders: erythema, flushes, skin and nail hyperpigmentation, photosensitivity, hypersensitivity to irradiated skin (radiation-recall reaction), urticaria
Renal and urinary disorders: red coloration of urine for 1 to 2 days after administration
General disorders and administration site conditions: fever, chills
Injury, poisoning and procedural complications: chemical cystitis (following intravesical administration)
8 Use in Specific Populations
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ELLENCE was evaluated in two studies (Studies MA-5 and GFEA-05) evaluating combination regimens in patients with early breast cancer [see Clinical Studies (14.1)]. Of the 1260 patients treated in these studies, 620 patients received the higher-dose ELLENCE regimen (FEC-100/CEF-120), 280 patients received the lower-dose ELLENCE regimen (FEC-50), and 360 patients received CMF. Serotonin-specific antiemetic therapy and colony-stimulating factors were not used in these trials. Clinically relevant adverse reactions are summarized in Table 1.
| Event | % of Patients | |||||
|---|---|---|---|---|---|---|
|
FEC-100/CEF-120
(N=620) |
FEC-50
(N=280) |
CMF
(N=360) |
||||
| Grades 1–4 | Grades 3/4 | Grades 1–4 | Grades 3/4 | Grades 1–4 | Grades 3/4 | |
| FEC & CEF = cyclophosphamide + ELLENCE + fluorouracil; CMF = cyclophosphamide + methotrexate + fluorouracil; NA = not available | ||||||
| Grade 1 or 2 changes in transaminase levels were observed but were more frequently seen with CMF than with CEF. | ||||||
|
Hematologic |
||||||
|
Leukopenia |
80 |
59 |
50 |
1.5 |
98 |
60 |
|
Neutropenia |
80 |
67 |
54 |
11 |
96 |
78 |
|
Anemia |
72 |
6 |
13 |
0 |
71 |
0.9 |
|
Thrombocytopenia |
49 |
5 |
4.6 |
0 |
51 |
3.6 |
|
Endocrine |
||||||
|
Amenorrhea |
72 |
0 |
69 |
0 |
68 |
0 |
|
Hot flashes |
39 |
4 |
5 |
0 |
69 |
6 |
|
Body as a Whole |
||||||
|
Lethargy |
46 |
1.9 |
1.1 |
0 |
73 |
0.3 |
|
Fever |
5 |
0 |
1.4 |
0 |
4.5 |
0 |
|
Gastrointestinal |
||||||
|
Nausea/vomiting |
92 |
25 |
83 |
22 |
85 |
6 |
|
Mucositis |
59 |
9 |
9 |
0 |
53 |
1.9 |
|
Diarrhea |
25 |
0.8 |
7 |
0 |
51 |
2.8 |
|
Anorexia |
2.9 |
0 |
1.8 |
0 |
6 |
0.3 |
|
Infection |
||||||
|
Infection |
22 |
1.6 |
15 |
0 |
26 |
0.6 |
|
Febrile neutropenia |
NA |
6 |
0 |
0 |
NA |
1.1 |
|
Ocular |
||||||
|
Conjunctivitis/keratitis |
15 |
0 |
1.1 |
0 |
38 |
0 |
|
Skin |
||||||
|
Alopecia |
96 |
57 |
70 |
19 |
84 |
7 |
|
Local toxicity |
20 |
0.3 |
2.5 |
0.4 |
8 |
0 |
|
Rash/itch |
9 |
0.3 |
1.4 |
0 |
14 |
0 |
|
Skin changes |
4.7 |
0 |
0.7 |
0 |
7 |
0 |
16 How Supplied/storage and Handling
ELLENCE is available in polypropylene single-dose CYTOSAFE® vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths:
|
Unit of Sale |
Strength |
|
NDC 0009-5091-01 Carton of 1 Single-dose Vial |
50 mg/25 mL (2 mg/mL) |
|
NDC 0009-5093-01 Carton of 1 Single-dose Vial |
200 mg/100 mL (2 mg/mL) |
Discard unused portion.
ELLENCE is available in ONCO-TAIN® glass vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths:
|
Unit of Sale |
Strength |
|
NDC 0009-5091-25 Carton of 1 Single-dose Vial |
50 mg/25 mL (2 mg/mL) |
|
NDC 0009-5093-10 Carton of 1 Single-dose Vial |
200 mg/100 mL (2 mg/mL) |
ONCO-TAIN® is the vial external protection system.
Discard unused portion.
5.3 Extravasation and Tissue Necrosis
Extravasation of ELLENCE can result in severe local tissue injury manifesting as blistering, ulceration, and necrosis requiring wide excision of the affected area and skin grafting. Extravasation should be considered if a patient experiences a burning or stinging sensation or shows other evidence indicating peri-venous infiltration or extravasation; however, extravasation may be present in patients who do not experience a stinging or burning sensation or when blood return is present on aspiration of the infusion needle.
Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Administer ELLENCE slowly into the tubing of a freely running intravenous infusion. Patients receiving initial therapy at the recommended starting doses of 100–120 mg/m2 should have ELLENCE infused over 15–20 minutes. For patients who require lower ELLENCE starting doses due to organ dysfunction or who require modification of ELLENCE doses during therapy, the ELLENCE infusion time may be proportionally decreased, but should not be less than 3 minutes [see Dosage and Administration (2.3)]. If possible, avoid veins over joints or in extremities with compromised venous or lymphatic drainage. Facial flushing, as well as local erythematous streaking along the vein, may be indicative of excessively rapid administration. It may precede local phlebitis or thrombophlebitis.
Immediately terminate infusion and restart in another vein if a burning or stinging sensation indicates perivenous infiltration. Perivenous infiltration may occur without causing pain. If extravasation is suspected, immediately discontinue the intravenous injection or continuous intravenous infusion [see Dosage and Administration (2.3)]. Apply ice to the site intermittently for 15 minutes, 4 times a day for 3 days. If appropriate, administer dexrazoxane at the site of extravasation as soon as possible and within the first 6 hours after extravasation.
14.1 Adjuvant Treatment of Breast Cancer
Two randomized, open-label, multicenter studies evaluated the use of ELLENCE 100 to 120 mg/m2 in combination with cyclophosphamide and fluorouracil for the adjuvant treatment of patients with axillary-node positive breast cancer and no evidence of distant metastatic disease (Stage II or III). Study MA-5 evaluated 120 mg/m2 of ELLENCE per course in combination with cyclophosphamide and fluorouracil (CEF-120 regimen). This study randomized premenopausal and perimenopausal women with one or more positive lymph nodes to an ELLENCE-containing CEF-120 regimen or to a CMF regimen. Study GFEA-05 evaluated the use of 100 mg/m2 of ELLENCE per course in combination with fluorouracil and cyclophosphamide (FEC-100). This study randomized pre- and postmenopausal women to the FEC-100 regimen or to a lower-dose FEC-50 regimen. In the GFEA-05 study, eligible patients were either required to have ≥ 4 nodes involved with tumor or, if only 1 to 3 nodes were positive, to have negative estrogen- and progesterone-receptors and a histologic tumor grade of 2 or 3. A total of 1281 women participated in these studies. Patients with T4 tumors were not eligible for either study. Table 5 shows the treatment regimens that the patients received. Relapse-free survival was defined as time to occurrence of a local, regional, or distant recurrence, or disease-related death. Patients with contralateral breast cancer, second primary malignancy, or death from causes other than breast cancer were censored at the time of the last visit prior to these events.
| Treatment Groups | Agent | Regimen | |
|---|---|---|---|
|
MA-5 In women who underwent lumpectomy, breast irradiation was to be administered after completion of study chemotherapy.
N=716 |
CEF-120 (total, 6 cycles) Patients also received prophylactic antibiotic therapy with trimethoprim-sulfamethoxazole or fluoroquinolone for the duration of their chemotherapy. N=356CMF (total, 6 cycles) N=360 |
Cyclophosphamide |
75 mg/m2 PO, d 1–14, q 28 days |
|
GFEA-05 All women were to receive breast irradiation after the completion of chemotherapy.
N=565 |
FEC-100 (total, 6 cycles) |
Fluorouracil |
500 mg/m2 IV, d 1, q 21 days |
In the MA-5 trial, the median age of the study population was 45 years. Approximately 60% of patients had 1 to 3 involved nodes and approximately 40% had ≥ 4 nodes involved with tumor. In the GFEA-05 study, the median age was 51 years and approximately half of the patients were postmenopausal. About 17% of the study population had 1 to 3 positive nodes and 80% of patients had ≥ 4 involved lymph nodes. Demographic and tumor characteristics were well-balanced between treatment arms in each study.
Relapse-free survival (RFS) and overall survival (OS) were analyzed using Kaplan-Meier methods in the intent-to-treat (ITT) patient populations in each study. Results were initially analyzed after up to 5 years of follow-up and these results are presented in the text below and in Table 6. Results after up to 10 years of follow-up are presented in Table 6. In Study MA-5, ELLENCE-containing combination therapy (CEF-120) showed significantly longer RFS than CMF (5-year estimates were 62% versus 53%, stratified logrank for the overall RFS p=0.013). The estimated reduction in the risk of relapse was 24% at 5 years. The OS was also greater for the ELLENCE-containing CEF-120 regimen than for the CMF regimen (5-year estimate 77% versus 70%; stratified logrank for overall survival p=0.043; non-stratified logrank p=0.13). The estimated reduction in the risk of death was 29% at 5 years.
In Study GFEA-05, patients treated with the higher-dose ELLENCE regimen (FEC-100) had a significantly longer 5-year RFS (estimated 65% versus 52%, logrank for the overall RFS p=0.007) and OS (estimated 76% versus 65%, logrank for the overall survival p=0.007) than patients given the lower dose regimen (FEC-50). The estimated reduction in risk of relapse was 32% at 5 years. The estimated reduction in the risk of death was 31% at 5 years. Results of follow-up up to 10 years (median follow-up = 8.8 years and 8.3 years, respectively, for Study MA-5 and Study GFEA-05) are presented in Table 6.
Although the trials were not powered for subgroup analyses, in the MA-5 study, improvements in favor of CEF-120 vs. CMF were observed, in RFS and OS both in patients with 1–3 node positive and in those with ≥4 node positive tumor involvement. In the GFEA-05 study, improvements in RFS and OS were observed in both pre-and postmenopausal women treated with FEC-100 compared to FEC-50.
| MA-5 Study | GFEA-05 Study | |||
|---|---|---|---|---|
| CEF-120 N=356 |
CMF N=360 |
FEC-100 N=276 |
FEC-50 N=289 |
|
|
RFS at 5 yrs (%) |
62 |
53 |
65 |
52 |
|
Hazard ratio Hazard ratio: CMF:CEF-120 in MA-5, FEC-50:FEC-100 in GFEA-05
|
0.76 |
0.68 |
||
|
2-sided 95% CI |
(0.60, 0.96) |
(0.52, 0.89) |
||
|
Logrank Test stratified Patients in MA-5 were stratified by nodal status (1–3, 4–10, and >10 positive nodes), type of initial surgery (lumpectomy versus mastectomy), and by hormone receptor status (ER or PR positive (≥10 fmol), both negative (<10 fmol), or unknown status). Patients in GFEA-05 were stratified by nodal status (1–3, 4–10, and >10 positive nodes).
|
(p = 0.013) |
(p = 0.007) |
||
|
OS at 5 yrs (%) |
77 |
70 |
76 |
65 |
|
Hazard ratio |
0.71 |
0.69 |
||
|
2-sided 95% CI |
(0.52, 0.98) |
(0.51, 0.92) |
||
|
Logrank Test stratified |
(p = 0.043) |
(p = 0.007) |
||
|
(unstratified p = 0.13) |
||||
|
RFS at 10 yrs (%) |
51 |
44 |
49 |
43 |
|
Hazard ratio |
0.78 |
0.78 |
||
|
2-sided 95% CI |
(0.63, 0.95) |
(0.62, 0.99) |
||
|
Logrank Test stratified |
(p = 0.017) |
(p = 0.040) |
||
|
(unstratified p = 0.023) |
(unstratified p = 0.09) |
|||
|
OS at 10 yrs (%) |
61 |
57 |
56 |
50 |
|
Hazard ratio |
0.82 |
0.75 |
||
|
2-sided 95% CI |
(0.65, 1.04) |
(0.58, 0.96) |
||
|
Logrank Test stratified |
(p = 0.100) |
(p = 0.023) |
||
|
(unstratified p = 0.18) |
(unstratified p = 0.039) |
The Kaplan-Meier curves for RFS and OS from Study MA-5 are shown in Figures 3 and 4 and those for Study GFEA-05 are shown in Figures 5 and 6.
Figure 3. Relapse-Free Survival in Study MA-5
Figure 4. Overall Survival in Study MA-5
Figure 5. Relapse-Free Survival in Study GFEA-05
Figure 6. Overall Survival in Study GFEA-05
2.1 Important Administration Instructions
When possible, to reduce the risk of developing cardiotoxicity in patients receiving ELLENCE after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, delay ELLENCE-based therapy until the other agents have cleared from the circulation [see Warnings and Precautions (5.1)].
Antiemetics may reduce nausea and vomiting; consider use of antiemetics before administration of ELLENCE or when clinically indicated, particularly when given in conjunction with other emetigenic drugs [see Adverse Reactions (6.1)].
Patients administered the 120 mg/m2 regimen of ELLENCE should receive prophylactic antibiotic therapy.
5.6 Use in Patients With Renal Impairment
Assess serum creatinine before and during therapy. Dosage adjustment is necessary in patients with serum creatinine >5 mg/dL [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]. Patients undergoing dialysis have not been studied.
5.5 Use in Patients With Hepatic Impairment
The major route of elimination of epirubicin is the hepatobiliary system [see Clinical Pharmacology (12.3)]. Evaluate serum total bilirubin and AST levels before and during treatment with ELLENCE. Patients with elevated bilirubin or AST may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients [see Dosage and Administration (2.2)]. Patients with severe hepatic impairment have not been evaluated; therefore, do not use ELLENCE in this patient population [see Contraindications (4)].
5.9 Thrombophlebitis and Thromboembolic Events
Thrombophlebitis and thromboembolic events, including pulmonary embolism (in some cases fatal) have been reported with the use of ELLENCE.
Principal Display Panel 50 Mg/25 Ml Vial Label
NDC 0009-5091-01
Rx only
Single-dose Vial
Ellence®
(epirubicin hydrochloride
injection)
50 mg/25 mL
(2 mg/mL)
For Intravenous Use Only
Principal Display Panel 50 Mg/25 Ml Vial Carton
NDC 0009-5091-01
Rx only
Single-dose Vial
Ellence®
(epirubicin hydrochloride
injection)
50 mg/25 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer Injectables
Principal Display Panel 200 Mg/100 Ml Vial Label
NDC 0009-5093-01
Rx only
Single-dose Vial
Ellence®
(epirubicin hydrochloride
injection)
200 mg/100 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer Injectables
Principal Display Panel 200 Mg/100 Ml Vial Carton
NDC 0009-5093-01
Rx only
Single-dose Vial
Ellence®
(epirubicin hydrochloride
injection)
200 mg/100 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer Injectables
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Conventional long-term animal studies to evaluate the carcinogenic potential of epirubicin have not been conducted, but intravenous administration of a single 3.6 mg/kg epirubicin dose to female rats (about 0.2 times the maximum recommended human dose on a body surface area basis) approximately doubled the incidence of mammary tumors (primarily fibroadenomas) observed at 1 year. Administration of 0.5 mg/kg epirubicin intravenously to rats (about 0.025 times the maximum recommended human dose on a body surface area basis) every 3 weeks for ten doses increased the incidence of subcutaneous fibromas in males over an 18-month observation period. In addition, subcutaneous administration of 0.75 or 1.0 mg/kg/day (about 0.015 times the maximum recommended human dose on a body surface area basis) to newborn rats for 4 days on both the first and tenth day after birth for a total of eight doses increased the incidence of animals with tumors compared to controls during a 24-month observation period.
Epirubicin was mutagenic in vitro to bacteria (Ames test) either in the presence or absence of metabolic activation and to mammalian cells (HGPRT assay in V79 Chinese hamster lung fibroblasts) in the absence but not in the presence of metabolic activation. Epirubicin was clastogenic in vitro (chromosome aberrations in human lymphocytes) both in the presence and absence of metabolic activation and was also clastogenic in vivo (chromosome aberration in mouse bone marrow).
In fertility studies in rats, males were given epirubicin daily for 9 weeks and mated with females that were given epirubicin daily for 2 weeks prior to mating and through Day 7 of gestation. When 0.3 mg/kg/day (about 0.015 times the maximum recommended human single dose on a body surface area basis) was administered to both sexes, no pregnancies resulted. No effects on mating behavior or fertility were observed at 0.1 mg/kg/day, but male rats had atrophy of the testes and epididymis, and reduced spermatogenesis. The 0.1 mg/kg/day dose also caused embryolethality. An increased incidence of fetal growth retardation was observed in these studies at 0.03 mg/kg/day (about 0.0015 times the maximum recommended human single dose on a body surface area basis). Multiple daily doses of epirubicin to rabbits and dogs also caused atrophy of male reproductive organs. Single 20.5 and 12 mg/kg doses of intravenous epirubicin caused testicular atrophy in mice and rats, respectively (both approximately 0.5 times the maximum recommended human dose on a body surface area basis). A single dose of 16.7 mg/kg epirubicin caused uterine atrophy in rats.
5.10 Potentiation of Radiation Toxicity and Radiation Recall
ELLENCE can increase radiation-induced toxicity to the myocardium, mucosa, skin, and liver. Radiation recall, including but not limited to cutaneous and pulmonary toxicity, can occur in patients who receive ELLENCE after prior radiation therapy.
Principal Display Panel 50 Mg/25 Ml Vial Label Onco Tain®
NDC 0009-5091-25
Rx only
Ellence®
(epirubicin hydrochloride
injection)
50 mg/25 mL
(2 mg/mL)
For Intravenous Use Only
Single-dose Vial.
Discard unused portion.
Principal Display Panel 50 Mg/25 Ml Vial Carton Onco Tain®
NDC 0009-5091-25
One 25 mL Single-dose Vial.
Discard unused portion.
Ellence®
(epirubicin hydrochloride
injection)
50 mg/25 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer
Hospital
Rx only
Warning: Hazardous Drug
Principal Display Panel 200 Mg/100 Ml Vial Label Onco Tain®
NDC 0009-5093-10
Rx only
Ellence®
(epirubicin hydrochloride injection)
200 mg/100 mL
(2 mg/mL)
For Intravenous Use Only
Single-dose Vial.
Discard unused portion.
Principal Display Panel 200 Mg/100 Ml Vial Carton Onco Tain®
NDC 0009-5093-10
One 100 mL Single-dose Vial.
Discard unused portion.
Ellence®
(epirubicin hydrochloride
injection)
200 mg/100 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer
Hospital
Rx only
Warning: Hazardous Drug
5.8 Immunosuppressant Effects/increased Susceptibility to Infections
Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including epirubicin, may result in serious or fatal infections. Avoid vaccination with a live vaccine in patients receiving ELLENCE. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
Warning: Cardiac Toxicity, Secondary Malignancies, Extravasation and Tissue Necrosis, and Severe Myelosuppression
-
•Cardiac Toxicity: Myocardial damage, including acute left ventricular failure, can occur with ELLENCE. The risk of cardiomyopathy is proportional to the cumulative exposure with incidence rates from 0.9% at a cumulative dose of 550 mg/m2, 1.6% at 700 mg/m2, and 3.3% at 900 mg/m2. The risk of cardiomyopathy is further increased with concomitant cardiotoxic therapy. Assess left ventricular ejection fraction (LVEF) before and regularly during and after treatment with ELLENCE [see Warnings and Precautions (5.1)].
-
•Secondary Malignancies: Secondary acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) occur at a higher incidence in patients treated with anthracyclines, including ELLENCE [see Warnings and Precautions (5.2)].
-
•Extravasation and Tissue Necrosis: Extravasation of ELLENCE can result in severe local tissue injury and necrosis requiring wide excision of the affected area and skin grafting. Immediately terminate the drug and apply ice to the affected area [see Warnings and Precautions (5.3)].
-
•Severe myelosuppression resulting in serious infection, septic shock, requirement for transfusions, hospitalization, and death may occur [see Warnings and Precautions (5.4)].
Structured Label Content
Section 42229-5 (42229-5)
Cardiac Toxicity
Discontinue ELLENCE in patients who develop signs or symptoms of cardiomyopathy [see Warnings and Precautions (5.1)].
Section 44425-7 (44425-7)
Store refrigerated between 2°C and 8°C (36°F and 46°F). Do not freeze. Protect from light.
Storage of the solution for injection at refrigerated conditions can result in the formation of a gelled product. This gelled product will return to a slightly viscous to mobile solution after 2 to a maximum of 4 hours equilibration at controlled room temperature (15–25°C). Solution for injection should be used within 24 hours after removal from refrigeration.
ELLENCE is a hazardous drug. Follow applicable special handling and disposal procedures1 [see References (15)].
10 Overdosage (10 OVERDOSAGE)
There is no known antidote for overdoses of ELLENCE. A 36-year-old man with non-Hodgkin's lymphoma received a daily 95 mg/m2 dose of ELLENCE for 5 consecutive days. Five days later, he developed bone marrow aplasia, grade 4 mucositis, and gastrointestinal bleeding. No signs of acute cardiac toxicity were observed. He was treated with antibiotics, colony-stimulating factors, and antifungal agents, and recovered completely. A 63-year-old woman with breast cancer and liver metastasis received a single 320 mg/m2 dose of ELLENCE. She was hospitalized with hyperthermia and developed multiple organ failure (respiratory and renal), with lactic acidosis, increased lactate dehydrogenase, and anuria. Death occurred within 24 hours after administration of ELLENCE. Additional instances of administration of doses higher than recommended have been reported at doses ranging from 150 to 250 mg/m2. The observed adverse events in these patients were qualitatively similar to known toxicities of epirubicin. Most of the patients recovered with appropriate supportive care.
If an overdose occurs, provide supportive treatment (including antibiotic therapy, blood and platelet transfusions, colony-stimulating factors, and intensive care as needed) until the recovery of toxicities. Delayed CHF has been observed months after anthracycline administration. Observe patients carefully over time for signs of CHF and provided with appropriate supportive therapy.
15 References (15 REFERENCES)
-
1."Hazardous Drugs". OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
11 Description (11 DESCRIPTION)
ELLENCE (epirubicin hydrochloride injection) is an anthracycline topoisomerase inhibitor for intravenous administration. ELLENCE is supplied as a sterile, clear, red solution and is available in polypropylene vials containing 50 and 200 mg of epirubicin hydrochloride as a preservative-free, ready-to-use solution. Each milliliter of solution contains 2 mg of epirubicin hydrochloride. Inactive ingredients include 9 mg sodium chloride, USP, and water for injection, USP. The pH of the solution has been adjusted to 3.0 with hydrochloric acid and/or sodium hydroxide, NF.
Epirubicin hydrochloride is the 4-epimer of doxorubicin and is a semi-synthetic derivative of daunorubicin. The chemical name is (8S- cis )-10-[(3-amino-2,3,6-trideoxy-α-L- arabino -hexopyranosyl)oxy]-7,8,9,10-tetrahydro6,8,11-trihydroxy-8-(hydroxyacetyl)-1-methoxy-5,12-naphthacenedione hydrochloride. The active ingredient is a red-orange hygroscopic powder, with the empirical formula C27 H29 NO11 HCl and a molecular weight of 579.95. The structural formula is as follows:
7.2 Cimetidine
Cimetidine increases the exposure to epirubicin [see Clinical Pharmacology (12.3)]. Discontinue cimetidine during treatment with ELLENCE.
8.4 Pediatric Use
Safety and effectiveness of ELLENCE have not been established in pediatric patients. Pediatric patients may be at greater risk for anthracycline-induced acute manifestations of cardiotoxicity or late cardiovascular dysfunction. The pharmacokinetics of epirubicin in pediatric patients have not been evaluated.
8.5 Geriatric Use
Clinical experience in patients who were 65 years of age and older who received ELLENCE chemotherapy regimens for primary breast cancer showed no overall differences in safety and effectiveness compared with younger patients.
In elderly female patients, closely monitor for increased toxicity due to the risk of decreased clearance of epirubicin [see Clinical Pharmacology (12.3)].
4 Contraindications (4 CONTRAINDICATIONS)
ELLENCE is contraindicated in patients with:
-
•Severe myocardial insufficiency [see Warnings and Precautions (5.1)]
-
•Recent myocardial infarction or severe arrhythmias, or previous treatment with maximum cumulative dose of anthracyclines [see Warnings and Precautions (5.1)]
-
•Severe persistent drug-induced myelosuppression [see Warnings and Precautions (5.4)]
-
•Severe hepatic impairment (defined as Child-Pugh Class C or serum bilirubin level greater than 5 mg/dL) [see Warnings and Precautions (5.5)]
-
•Severe hypersensitivity to ELLENCE, other anthracyclines, or anthracenediones [see Adverse Reactions (6.1)]
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following clinically significant adverse reactions are described elsewhere in the labeling:
-
•Cardiac Toxicity [see Warnings and Precautions (5.1)]
-
•Secondary Malignancies [see Warnings and Precautions (5.2)]
-
•Extravasation and Tissue Necrosis [see Warnings and Precautions (5.3)]
-
•Severe Myelosuppression [see Warnings and Precautions (5.4)]
-
•Tumor-Lysis Syndrome [see Warnings and Precautions (5.7)]
-
•Thrombophlebitis and Thromboembolic Events [see Warnings and Precautions (5.9)]
-
•Potentiation of Radiation Toxicity and Radiation Recall [see Warnings and Precautions (5.10)]
7 Drug Interactions (7 DRUG INTERACTIONS)
2.2 Recommended Dose
The recommended dose of ELLENCE is 100 to 120 mg/m2 administered as an intravenous bolus [see Dosage and Administration (2.4)].
The following regimens are recommended:
|
CEF-120: |
Cyclophosphamide |
75 mg/m2 oral on Days 1 to 14 |
|
ELLENCE |
60 mg/m2 intravenously on Days 1 and 8 |
|
|
5-Fluorouracil |
500 mg/m2 intravenously on Days 1 and 8 |
|
|
Repeat every 28 days for 6 cycles |
||
|
FEC-100: |
5-Fluorouracil |
500 mg/m2 intravenously on Day 1 |
|
ELLENCE |
100 mg/m2 intravenously on Day 1 |
|
|
Cyclophosphamide |
500 mg/m2 intravenously on Day 1 |
|
|
Repeat every 21 days for 6 cycles |
Administer ELLENCE in repeated 3- to 4-week cycles. The total dose of ELLENCE may be given on Day 1 of each cycle or divided equally and given on Days 1 and 8 of each cycle.
5.1 Cardiac Toxicity
ELLENCE and other anthracycline drugs can result in either early (or acute) or late (delayed) cardiac toxicity.
The principal manifestations of early cardiac toxicity are sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes. However, tachycardia (including premature ventricular contractions and ventricular tachycardia), bradycardia, as well as atrioventricular and bundle-branch block have been reported. Early cardiac toxicity does not usually predict the subsequent occurrence of delayed cardiotoxicity and generally should not be considered a reason for suspending treatment with ELLENCE.
Delayed cardiac toxicity is manifested by reduced left ventricular ejection fraction (LVEF) and/or signs and symptoms of congestive heart failure (CHF). If it occurs, late cardiotoxicity usually develops late during therapy with ELLENCE or within 2 to 3 months after completion of treatment, but there are reports of it occurring several months to years after treatment termination. In a retrospective survey, including 9144 patients, mostly with solid tumors in advanced stages, the probability of developing CHF increased with increasing cumulative doses of ELLENCE (Figure 1). In another retrospective survey of 469 ELLENCE-treated patients with metastatic or early breast cancer, the reported risk of CHF was comparable to that observed in the larger study of over 9000 patients.
Given the risk of cardiac toxicity, cumulative doses of 900 mg/m2 ELLENCE should generally be avoided.
Figure 1. Risk of CHF in 9144 Patients Treated with ELLENCE
Prior history of cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, and concomitant use of other cardiotoxic drugs, increase the risk of developing late cardiac toxicity. Avoid administration of ELLENCE in combination with other cardiotoxic drugs. Although not formally tested, it is probable that the toxicity of ELLENCE and other anthracyclines or anthracenediones is additive. Cardiac toxicity with ELLENCE may occur at lower cumulative doses whether or not cardiac risk factors are present. Patients receiving ELLENCE after stopping treatment with other cardiotoxic drugs, especially those with long half-lives such as trastuzumab, may be at increased risk of developing cardiotoxicity [see Dosage and Administration (2) and Drug Interaction (7.1)].
Perform a baseline ECG and evaluation of LVEF prior to initiating treatment with ELLENCE. Monitor LVEF during the course of treatment and consider discontinuation of ELLENCE if LVEF decrease and/or signs or symptoms of CHF develop. Closely monitor patients with other risk factors for cardiac toxicity, particularly prior administration of anthracycline or anthracenedione.
8.7 Renal Impairment
No significant alterations in the pharmacokinetics of epirubicin or its major metabolite, epirubicinol, have been observed in patients with serum creatinine <5 mg/dL. Consider lower doses in patients with severe renal impairment (serum creatinine >5 mg/dL), as a reduction in plasma clearance was reported in these patients [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)]. Patients on dialysis have not been studied.
12.3 Pharmacokinetics
Epirubicin pharmacokinetics are linear over the dose range of 60 to 150 mg/m2 and plasma clearance is not affected by the duration of infusion or administration schedule. Pharmacokinetic parameters for epirubicin following 6- to 10-minute, single-dose intravenous infusions of ELLENCE at doses of 60 to 150 mg/m2 in patients with solid tumors are shown in Table 4. The plasma concentration declined in a triphasic manner with mean half-lives for the alpha, beta, and gamma phases of about 3 minutes, 2.5 hours, and 33 hours, respectively.
|
Dose
N=6 patients per dose level
(mg/m2) |
Cmax
Plasma concentration at the end of 6 to 10 minutes infusion
(µg/mL) |
AUC
Area under the plasma concentration curve
(µg∙h/mL) |
t1/2
Half-life of terminal phase
(hours) |
CL
Plasma clearance
(L/hour) |
Vss
Steady state volume of distribution
(L/kg) |
|---|---|---|---|---|---|
|
60 |
5.7 ± 1.6 |
1.6 ± 0.2 |
35.3 ± 9 |
65 ± 8 |
21 ± 2 |
|
75 |
5.3 ± 1.5 |
1.7 ± 0.3 |
32.1 ± 5 |
83 ± 14 |
27 ± 11 |
|
120 |
9.0 ± 3.5 |
3.4 ± 0.7 |
33.7 ± 4 |
65 ± 13 |
23 ± 7 |
|
150 |
9.3 ± 2.9 |
4.2 ± 0.8 |
31.1 ± 6 |
69 ± 13 |
21 ± 7 |
7.4 Radiation Therapy
There are few data regarding the coadministration of radiation therapy and ELLENCE. In adjuvant trials of ELLENCE-containing CEF-120 or FEC-100 chemotherapies, breast irradiation was delayed until after chemotherapy was completed. This practice resulted in no apparent increase in local breast cancer recurrence relative to published accounts in the literature. A small number of patients received ELLENCE-based chemotherapy concomitantly with radiation therapy but had chemotherapy interrupted in order to avoid potential overlapping toxicities. It is likely that use of ELLENCE with radiotherapy may sensitize tissues to the cytotoxic actions of irradiation. Administration of ELLENCE after previous radiation therapy may induce an inflammatory recall reaction at the site of the irradiation.
2.3 Dose Modifications
ELLENCE dosage adjustments for hematologic and non-hematologic toxicities within a cycle of treatment, is based on nadir platelet counts <50,000/mm3, absolute neutrophil counts (ANC) <250/mm3, neutropenic fever, or Grades 3/4 nonhematologic toxicity. Reduce ELLENCE Day 1 dose in subsequent cycles to 75% of the Day 1 dose given in the current cycle. Delay Day 1 chemotherapy in subsequent courses of treatment until platelet counts are ≥100,000/mm3, ANC ≥1500/mm3, and nonhematologic toxicities have recovered to ≤ Grade 1.
7.1 Cardiotoxic Agents
Closely monitor cardiac function when ELLENCE is used in combination with other cardiotoxic agents. Patients receiving ELLENCE after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, may be at an increased risk of developing cardiotoxicity [see Dosage and Administration (2) and Warnings and Precautions (5.1)]. Trastuzumab may persist in the circulation for up to 7 months. Therefore, avoid anthracycline-based therapy for up to 7 months after stopping trastuzumab when possible. Monitor the patient's cardiac function closely if anthracyclines are used before this time.
Concomitant use of ELLENCE with other cardioactive compounds that could cause heart failure (e.g., calcium channel blockers), requires close monitoring of cardiac function throughout treatment.
8.6 Hepatic Impairment
Epirubicin is eliminated by both hepatic metabolism and biliary excretion and clearance is reduced in patients with hepatic dysfunction. Do not treat patients with severe hepatic impairment with ELLENCE [see Contraindications (4)]. Reduce the starting dose for patients with less severe hepatic impairment [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
1 Indications and Usage (1 INDICATIONS AND USAGE)
ELLENCE is indicated as a component of adjuvant therapy in patients with evidence of axillary node tumor involvement following resection of primary breast cancer [see Clinical Studies (14.1)].
12.1 Mechanism of Action
Epirubicin is an anthracycline cytotoxic agent. Although it is known that anthracyclines can interfere with a number of biochemical and biological functions within eukaryotic cells, the precise mechanisms of epirubicin's cytotoxic and/or antiproliferative properties have not been completely elucidated.
Epirubicin forms a complex with DNA by intercalation of its planar rings between nucleotide base pairs, with consequent inhibition of nucleic acid (DNA and RNA) and protein synthesis.
Such intercalation triggers DNA cleavage by topoisomerase II, resulting in cytocidal activity. Epirubicin also inhibits DNA helicase activity, preventing the enzymatic separation of double-stranded DNA and interfering with replication and transcription. Epirubicin is also involved in oxidation/reduction reactions by generating cytotoxic free radicals. The antiproliferative and cytotoxic activity of epirubicin is thought to result from these or other possible mechanisms.
Epirubicin is cytotoxic in vitro to a variety of established murine and human cell lines and primary cultures of human tumors. It is also active in vivo against a variety of murine tumors and human xenografts in athymic mice, including breast tumors.
5.7 Tumor Lysis Syndrome (5.7 Tumor-Lysis Syndrome)
ELLENCE can induce tumor lysis syndrome in patients with rapidly growing tumors. Evaluate blood uric acid levels, potassium, calcium, phosphate, and creatinine after initial treatment. Consider hydration, urine alkalinization, and prophylaxis with allopurinol to minimize hyperuricemia and potential complications of tumor lysis syndrome.
7.3 Other Cytotoxic Drugs
ELLENCE used in combination with other cytotoxic drugs may show on-treatment additive toxicity, especially hematologic and gastrointestinal effects.
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
-
•Use in Patients with Hepatic Impairment: Monitor serum total bilirubin and AST levels before and during treatment with ELLENCE. In patients with elevated serum AST or serum total bilirubin, dosage reductions or discontinuation may be required (2.3, 5.5).
-
•Tumor Lysis Syndrome: Evaluate blood uric acid levels, potassium, calcium, phosphate, and creatinine after initial treatment. Consider hydration, urine alkalinization, and prophylaxis with allopurinol to minimize potential complications of hyperuricemia and tumor lysis syndrome (5.7).
-
•Thrombophlebitis and Thromboembolic Events: Thrombophlebitis and thromboembolic events, including pulmonary embolism (in some cases fatal) have been reported with the use of ELLENCE. Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein (5.9).
-
•Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including ELLENCE, may result in serious or fatal infections (5.7).
-
•Potentiation of Radiation Toxicity and Radiation Recall: Administration of ELLENCE after previous radiation therapy may induce an inflammatory recall reaction at the site of the irradiation (5.10).
-
•Embryo-Fetal Toxicity: ELLENCE can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception (5.11, 8.1, 8.3).
5.11 Embryo Fetal Toxicity (5.11 Embryo-Fetal Toxicity)
Based on findings from animals and its mechanism of action, ELLENCE can cause fetal harm when administered to a pregnant woman; avoid the use of ELLENCE during the 1st trimester. Available human data do not establish the presence or absence of major birth defects and miscarriage related to the use of epirubicin during the 2nd and 3rd trimesters. In animal reproduction studies, epirubicin was embryo-fetal lethal and caused structural abnormalities in rats and rabbits at doses less than the maximum recommended human dose on a body surface area basis. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise female patients of reproductive potential to use effective contraception during treatment and for 6 months after the last dose of ELLENCE. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ELLENCE. Advise male patients with pregnant partners to use condoms during treatment and for at least 7 days after the last dose of ELLENCE [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1), and Nonclinical Toxicology (13.1)].
5.2 Secondary Malignancies
The risk of developing secondary acute myelogenous leukemia and myelodysplastic syndrome (MDS), is increased following treatment with ELLENCE and other anthracyclines. Cumulative risk of secondary acute myelogenous leukemia or myelodysplastic syndrome (AML/MDS) of about 0.27% at 3 years, 0.46% at 5 years, and 0.55% at 8 years. These leukemias generally occur within 1 to 3 years of treatment [see Adverse Reactions (6.1)].
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
-
•The recommended starting dose of ELLENCE is 100 to 120 mg/m2. Dosage reductions are possible when given in certain combinations (2.2).
-
•Administer intravenously in repeated 3- to 4-week cycles, either total dose on Day 1 of each cycle or divided equally and given on Days 1 and 8 of each cycle (2.2).
-
•Consider use of antiemetics when given in conjunction with other emetigenic drugs (2.1).
-
•Patients administered the 120 mg/m2 regimen of ELLENCE should receive prophylactic antibiotic therapy (2.1).
-
•Adjust dosage after the first treatment cycle based on hematologic and nonhematologic toxicities (2.3).
-
•Reduce dose in patients with hepatic impairment (2.3, 8.6).
-
•Consider lower doses in patients with severe renal impairment (2.3, 8.7).
5.4 Severe Myelosuppression
ELLENCE can cause severe myelosuppression [see Adverse Reactions (6.1) . Obtain complete blood counts prior to each treatment and carefully monitor patients during treatment for possible clinical complications due to myelosuppression. Delay the next dose of ELLENCE if severe myelosuppression has not improved. Consider dose reduction for patients with prolonged myelosuppression based on the severity of reaction [see Dosage and Administration (2.3)].
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
Injection: 50 mg/25 mL (2 mg/mL), 200 mg/100 mL (2 mg/mL) clear red solution in a single-dose vial.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ELLENCE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Infections and infestations: sepsis, pneumonia
Immune system disorders: anaphylaxis
Metabolism and nutrition disorders: dehydration, hyperuricemia
Vascular disorders: shock, hemorrhage, embolism arterial, thrombophlebitis, phlebitis
Respiratory, thoracic and mediastinal disorders: pulmonary embolism
Gastrointestinal disorders: erosions, ulcerations, pain or burning sensation, bleeding, hyperpigmentation of the oral mucosa
Skin and subcutaneous tissue disorders: erythema, flushes, skin and nail hyperpigmentation, photosensitivity, hypersensitivity to irradiated skin (radiation-recall reaction), urticaria
Renal and urinary disorders: red coloration of urine for 1 to 2 days after administration
General disorders and administration site conditions: fever, chills
Injury, poisoning and procedural complications: chemical cystitis (following intravesical administration)
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ELLENCE was evaluated in two studies (Studies MA-5 and GFEA-05) evaluating combination regimens in patients with early breast cancer [see Clinical Studies (14.1)]. Of the 1260 patients treated in these studies, 620 patients received the higher-dose ELLENCE regimen (FEC-100/CEF-120), 280 patients received the lower-dose ELLENCE regimen (FEC-50), and 360 patients received CMF. Serotonin-specific antiemetic therapy and colony-stimulating factors were not used in these trials. Clinically relevant adverse reactions are summarized in Table 1.
| Event | % of Patients | |||||
|---|---|---|---|---|---|---|
|
FEC-100/CEF-120
(N=620) |
FEC-50
(N=280) |
CMF
(N=360) |
||||
| Grades 1–4 | Grades 3/4 | Grades 1–4 | Grades 3/4 | Grades 1–4 | Grades 3/4 | |
| FEC & CEF = cyclophosphamide + ELLENCE + fluorouracil; CMF = cyclophosphamide + methotrexate + fluorouracil; NA = not available | ||||||
| Grade 1 or 2 changes in transaminase levels were observed but were more frequently seen with CMF than with CEF. | ||||||
|
Hematologic |
||||||
|
Leukopenia |
80 |
59 |
50 |
1.5 |
98 |
60 |
|
Neutropenia |
80 |
67 |
54 |
11 |
96 |
78 |
|
Anemia |
72 |
6 |
13 |
0 |
71 |
0.9 |
|
Thrombocytopenia |
49 |
5 |
4.6 |
0 |
51 |
3.6 |
|
Endocrine |
||||||
|
Amenorrhea |
72 |
0 |
69 |
0 |
68 |
0 |
|
Hot flashes |
39 |
4 |
5 |
0 |
69 |
6 |
|
Body as a Whole |
||||||
|
Lethargy |
46 |
1.9 |
1.1 |
0 |
73 |
0.3 |
|
Fever |
5 |
0 |
1.4 |
0 |
4.5 |
0 |
|
Gastrointestinal |
||||||
|
Nausea/vomiting |
92 |
25 |
83 |
22 |
85 |
6 |
|
Mucositis |
59 |
9 |
9 |
0 |
53 |
1.9 |
|
Diarrhea |
25 |
0.8 |
7 |
0 |
51 |
2.8 |
|
Anorexia |
2.9 |
0 |
1.8 |
0 |
6 |
0.3 |
|
Infection |
||||||
|
Infection |
22 |
1.6 |
15 |
0 |
26 |
0.6 |
|
Febrile neutropenia |
NA |
6 |
0 |
0 |
NA |
1.1 |
|
Ocular |
||||||
|
Conjunctivitis/keratitis |
15 |
0 |
1.1 |
0 |
38 |
0 |
|
Skin |
||||||
|
Alopecia |
96 |
57 |
70 |
19 |
84 |
7 |
|
Local toxicity |
20 |
0.3 |
2.5 |
0.4 |
8 |
0 |
|
Rash/itch |
9 |
0.3 |
1.4 |
0 |
14 |
0 |
|
Skin changes |
4.7 |
0 |
0.7 |
0 |
7 |
0 |
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
ELLENCE is available in polypropylene single-dose CYTOSAFE® vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths:
|
Unit of Sale |
Strength |
|
NDC 0009-5091-01 Carton of 1 Single-dose Vial |
50 mg/25 mL (2 mg/mL) |
|
NDC 0009-5093-01 Carton of 1 Single-dose Vial |
200 mg/100 mL (2 mg/mL) |
Discard unused portion.
ELLENCE is available in ONCO-TAIN® glass vials containing 2 mg epirubicin hydrochloride per mL as a sterile, preservative-free, ready-to-use, clear, red solution in the following strengths:
|
Unit of Sale |
Strength |
|
NDC 0009-5091-25 Carton of 1 Single-dose Vial |
50 mg/25 mL (2 mg/mL) |
|
NDC 0009-5093-10 Carton of 1 Single-dose Vial |
200 mg/100 mL (2 mg/mL) |
ONCO-TAIN® is the vial external protection system.
Discard unused portion.
5.3 Extravasation and Tissue Necrosis
Extravasation of ELLENCE can result in severe local tissue injury manifesting as blistering, ulceration, and necrosis requiring wide excision of the affected area and skin grafting. Extravasation should be considered if a patient experiences a burning or stinging sensation or shows other evidence indicating peri-venous infiltration or extravasation; however, extravasation may be present in patients who do not experience a stinging or burning sensation or when blood return is present on aspiration of the infusion needle.
Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Administer ELLENCE slowly into the tubing of a freely running intravenous infusion. Patients receiving initial therapy at the recommended starting doses of 100–120 mg/m2 should have ELLENCE infused over 15–20 minutes. For patients who require lower ELLENCE starting doses due to organ dysfunction or who require modification of ELLENCE doses during therapy, the ELLENCE infusion time may be proportionally decreased, but should not be less than 3 minutes [see Dosage and Administration (2.3)]. If possible, avoid veins over joints or in extremities with compromised venous or lymphatic drainage. Facial flushing, as well as local erythematous streaking along the vein, may be indicative of excessively rapid administration. It may precede local phlebitis or thrombophlebitis.
Immediately terminate infusion and restart in another vein if a burning or stinging sensation indicates perivenous infiltration. Perivenous infiltration may occur without causing pain. If extravasation is suspected, immediately discontinue the intravenous injection or continuous intravenous infusion [see Dosage and Administration (2.3)]. Apply ice to the site intermittently for 15 minutes, 4 times a day for 3 days. If appropriate, administer dexrazoxane at the site of extravasation as soon as possible and within the first 6 hours after extravasation.
14.1 Adjuvant Treatment of Breast Cancer
Two randomized, open-label, multicenter studies evaluated the use of ELLENCE 100 to 120 mg/m2 in combination with cyclophosphamide and fluorouracil for the adjuvant treatment of patients with axillary-node positive breast cancer and no evidence of distant metastatic disease (Stage II or III). Study MA-5 evaluated 120 mg/m2 of ELLENCE per course in combination with cyclophosphamide and fluorouracil (CEF-120 regimen). This study randomized premenopausal and perimenopausal women with one or more positive lymph nodes to an ELLENCE-containing CEF-120 regimen or to a CMF regimen. Study GFEA-05 evaluated the use of 100 mg/m2 of ELLENCE per course in combination with fluorouracil and cyclophosphamide (FEC-100). This study randomized pre- and postmenopausal women to the FEC-100 regimen or to a lower-dose FEC-50 regimen. In the GFEA-05 study, eligible patients were either required to have ≥ 4 nodes involved with tumor or, if only 1 to 3 nodes were positive, to have negative estrogen- and progesterone-receptors and a histologic tumor grade of 2 or 3. A total of 1281 women participated in these studies. Patients with T4 tumors were not eligible for either study. Table 5 shows the treatment regimens that the patients received. Relapse-free survival was defined as time to occurrence of a local, regional, or distant recurrence, or disease-related death. Patients with contralateral breast cancer, second primary malignancy, or death from causes other than breast cancer were censored at the time of the last visit prior to these events.
| Treatment Groups | Agent | Regimen | |
|---|---|---|---|
|
MA-5 In women who underwent lumpectomy, breast irradiation was to be administered after completion of study chemotherapy.
N=716 |
CEF-120 (total, 6 cycles) Patients also received prophylactic antibiotic therapy with trimethoprim-sulfamethoxazole or fluoroquinolone for the duration of their chemotherapy. N=356CMF (total, 6 cycles) N=360 |
Cyclophosphamide |
75 mg/m2 PO, d 1–14, q 28 days |
|
GFEA-05 All women were to receive breast irradiation after the completion of chemotherapy.
N=565 |
FEC-100 (total, 6 cycles) |
Fluorouracil |
500 mg/m2 IV, d 1, q 21 days |
In the MA-5 trial, the median age of the study population was 45 years. Approximately 60% of patients had 1 to 3 involved nodes and approximately 40% had ≥ 4 nodes involved with tumor. In the GFEA-05 study, the median age was 51 years and approximately half of the patients were postmenopausal. About 17% of the study population had 1 to 3 positive nodes and 80% of patients had ≥ 4 involved lymph nodes. Demographic and tumor characteristics were well-balanced between treatment arms in each study.
Relapse-free survival (RFS) and overall survival (OS) were analyzed using Kaplan-Meier methods in the intent-to-treat (ITT) patient populations in each study. Results were initially analyzed after up to 5 years of follow-up and these results are presented in the text below and in Table 6. Results after up to 10 years of follow-up are presented in Table 6. In Study MA-5, ELLENCE-containing combination therapy (CEF-120) showed significantly longer RFS than CMF (5-year estimates were 62% versus 53%, stratified logrank for the overall RFS p=0.013). The estimated reduction in the risk of relapse was 24% at 5 years. The OS was also greater for the ELLENCE-containing CEF-120 regimen than for the CMF regimen (5-year estimate 77% versus 70%; stratified logrank for overall survival p=0.043; non-stratified logrank p=0.13). The estimated reduction in the risk of death was 29% at 5 years.
In Study GFEA-05, patients treated with the higher-dose ELLENCE regimen (FEC-100) had a significantly longer 5-year RFS (estimated 65% versus 52%, logrank for the overall RFS p=0.007) and OS (estimated 76% versus 65%, logrank for the overall survival p=0.007) than patients given the lower dose regimen (FEC-50). The estimated reduction in risk of relapse was 32% at 5 years. The estimated reduction in the risk of death was 31% at 5 years. Results of follow-up up to 10 years (median follow-up = 8.8 years and 8.3 years, respectively, for Study MA-5 and Study GFEA-05) are presented in Table 6.
Although the trials were not powered for subgroup analyses, in the MA-5 study, improvements in favor of CEF-120 vs. CMF were observed, in RFS and OS both in patients with 1–3 node positive and in those with ≥4 node positive tumor involvement. In the GFEA-05 study, improvements in RFS and OS were observed in both pre-and postmenopausal women treated with FEC-100 compared to FEC-50.
| MA-5 Study | GFEA-05 Study | |||
|---|---|---|---|---|
| CEF-120 N=356 |
CMF N=360 |
FEC-100 N=276 |
FEC-50 N=289 |
|
|
RFS at 5 yrs (%) |
62 |
53 |
65 |
52 |
|
Hazard ratio Hazard ratio: CMF:CEF-120 in MA-5, FEC-50:FEC-100 in GFEA-05
|
0.76 |
0.68 |
||
|
2-sided 95% CI |
(0.60, 0.96) |
(0.52, 0.89) |
||
|
Logrank Test stratified Patients in MA-5 were stratified by nodal status (1–3, 4–10, and >10 positive nodes), type of initial surgery (lumpectomy versus mastectomy), and by hormone receptor status (ER or PR positive (≥10 fmol), both negative (<10 fmol), or unknown status). Patients in GFEA-05 were stratified by nodal status (1–3, 4–10, and >10 positive nodes).
|
(p = 0.013) |
(p = 0.007) |
||
|
OS at 5 yrs (%) |
77 |
70 |
76 |
65 |
|
Hazard ratio |
0.71 |
0.69 |
||
|
2-sided 95% CI |
(0.52, 0.98) |
(0.51, 0.92) |
||
|
Logrank Test stratified |
(p = 0.043) |
(p = 0.007) |
||
|
(unstratified p = 0.13) |
||||
|
RFS at 10 yrs (%) |
51 |
44 |
49 |
43 |
|
Hazard ratio |
0.78 |
0.78 |
||
|
2-sided 95% CI |
(0.63, 0.95) |
(0.62, 0.99) |
||
|
Logrank Test stratified |
(p = 0.017) |
(p = 0.040) |
||
|
(unstratified p = 0.023) |
(unstratified p = 0.09) |
|||
|
OS at 10 yrs (%) |
61 |
57 |
56 |
50 |
|
Hazard ratio |
0.82 |
0.75 |
||
|
2-sided 95% CI |
(0.65, 1.04) |
(0.58, 0.96) |
||
|
Logrank Test stratified |
(p = 0.100) |
(p = 0.023) |
||
|
(unstratified p = 0.18) |
(unstratified p = 0.039) |
The Kaplan-Meier curves for RFS and OS from Study MA-5 are shown in Figures 3 and 4 and those for Study GFEA-05 are shown in Figures 5 and 6.
Figure 3. Relapse-Free Survival in Study MA-5
Figure 4. Overall Survival in Study MA-5
Figure 5. Relapse-Free Survival in Study GFEA-05
Figure 6. Overall Survival in Study GFEA-05
2.1 Important Administration Instructions
When possible, to reduce the risk of developing cardiotoxicity in patients receiving ELLENCE after stopping treatment with other cardiotoxic agents, especially those with long half-lives such as trastuzumab, delay ELLENCE-based therapy until the other agents have cleared from the circulation [see Warnings and Precautions (5.1)].
Antiemetics may reduce nausea and vomiting; consider use of antiemetics before administration of ELLENCE or when clinically indicated, particularly when given in conjunction with other emetigenic drugs [see Adverse Reactions (6.1)].
Patients administered the 120 mg/m2 regimen of ELLENCE should receive prophylactic antibiotic therapy.
5.6 Use in Patients With Renal Impairment (5.6 Use in Patients with Renal Impairment)
Assess serum creatinine before and during therapy. Dosage adjustment is necessary in patients with serum creatinine >5 mg/dL [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]. Patients undergoing dialysis have not been studied.
5.5 Use in Patients With Hepatic Impairment (5.5 Use in Patients with Hepatic Impairment)
The major route of elimination of epirubicin is the hepatobiliary system [see Clinical Pharmacology (12.3)]. Evaluate serum total bilirubin and AST levels before and during treatment with ELLENCE. Patients with elevated bilirubin or AST may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients [see Dosage and Administration (2.2)]. Patients with severe hepatic impairment have not been evaluated; therefore, do not use ELLENCE in this patient population [see Contraindications (4)].
5.9 Thrombophlebitis and Thromboembolic Events
Thrombophlebitis and thromboembolic events, including pulmonary embolism (in some cases fatal) have been reported with the use of ELLENCE.
Principal Display Panel 50 Mg/25 Ml Vial Label (PRINCIPAL DISPLAY PANEL - 50 mg/25 mL Vial Label)
NDC 0009-5091-01
Rx only
Single-dose Vial
Ellence®
(epirubicin hydrochloride
injection)
50 mg/25 mL
(2 mg/mL)
For Intravenous Use Only
Principal Display Panel 50 Mg/25 Ml Vial Carton (PRINCIPAL DISPLAY PANEL - 50 mg/25 mL Vial Carton)
NDC 0009-5091-01
Rx only
Single-dose Vial
Ellence®
(epirubicin hydrochloride
injection)
50 mg/25 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer Injectables
Principal Display Panel 200 Mg/100 Ml Vial Label (PRINCIPAL DISPLAY PANEL - 200 mg/100 mL Vial Label)
NDC 0009-5093-01
Rx only
Single-dose Vial
Ellence®
(epirubicin hydrochloride
injection)
200 mg/100 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer Injectables
Principal Display Panel 200 Mg/100 Ml Vial Carton (PRINCIPAL DISPLAY PANEL - 200 mg/100 mL Vial Carton)
NDC 0009-5093-01
Rx only
Single-dose Vial
Ellence®
(epirubicin hydrochloride
injection)
200 mg/100 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer Injectables
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Conventional long-term animal studies to evaluate the carcinogenic potential of epirubicin have not been conducted, but intravenous administration of a single 3.6 mg/kg epirubicin dose to female rats (about 0.2 times the maximum recommended human dose on a body surface area basis) approximately doubled the incidence of mammary tumors (primarily fibroadenomas) observed at 1 year. Administration of 0.5 mg/kg epirubicin intravenously to rats (about 0.025 times the maximum recommended human dose on a body surface area basis) every 3 weeks for ten doses increased the incidence of subcutaneous fibromas in males over an 18-month observation period. In addition, subcutaneous administration of 0.75 or 1.0 mg/kg/day (about 0.015 times the maximum recommended human dose on a body surface area basis) to newborn rats for 4 days on both the first and tenth day after birth for a total of eight doses increased the incidence of animals with tumors compared to controls during a 24-month observation period.
Epirubicin was mutagenic in vitro to bacteria (Ames test) either in the presence or absence of metabolic activation and to mammalian cells (HGPRT assay in V79 Chinese hamster lung fibroblasts) in the absence but not in the presence of metabolic activation. Epirubicin was clastogenic in vitro (chromosome aberrations in human lymphocytes) both in the presence and absence of metabolic activation and was also clastogenic in vivo (chromosome aberration in mouse bone marrow).
In fertility studies in rats, males were given epirubicin daily for 9 weeks and mated with females that were given epirubicin daily for 2 weeks prior to mating and through Day 7 of gestation. When 0.3 mg/kg/day (about 0.015 times the maximum recommended human single dose on a body surface area basis) was administered to both sexes, no pregnancies resulted. No effects on mating behavior or fertility were observed at 0.1 mg/kg/day, but male rats had atrophy of the testes and epididymis, and reduced spermatogenesis. The 0.1 mg/kg/day dose also caused embryolethality. An increased incidence of fetal growth retardation was observed in these studies at 0.03 mg/kg/day (about 0.0015 times the maximum recommended human single dose on a body surface area basis). Multiple daily doses of epirubicin to rabbits and dogs also caused atrophy of male reproductive organs. Single 20.5 and 12 mg/kg doses of intravenous epirubicin caused testicular atrophy in mice and rats, respectively (both approximately 0.5 times the maximum recommended human dose on a body surface area basis). A single dose of 16.7 mg/kg epirubicin caused uterine atrophy in rats.
5.10 Potentiation of Radiation Toxicity and Radiation Recall
ELLENCE can increase radiation-induced toxicity to the myocardium, mucosa, skin, and liver. Radiation recall, including but not limited to cutaneous and pulmonary toxicity, can occur in patients who receive ELLENCE after prior radiation therapy.
Principal Display Panel 50 Mg/25 Ml Vial Label Onco Tain® (PRINCIPAL DISPLAY PANEL - 50 mg/25 mL Vial Label ONCO-TAIN®)
NDC 0009-5091-25
Rx only
Ellence®
(epirubicin hydrochloride
injection)
50 mg/25 mL
(2 mg/mL)
For Intravenous Use Only
Single-dose Vial.
Discard unused portion.
Principal Display Panel 50 Mg/25 Ml Vial Carton Onco Tain® (PRINCIPAL DISPLAY PANEL - 50 mg/25 mL Vial Carton ONCO-TAIN®)
NDC 0009-5091-25
One 25 mL Single-dose Vial.
Discard unused portion.
Ellence®
(epirubicin hydrochloride
injection)
50 mg/25 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer
Hospital
Rx only
Warning: Hazardous Drug
Principal Display Panel 200 Mg/100 Ml Vial Label Onco Tain® (PRINCIPAL DISPLAY PANEL - 200 mg/100 mL Vial Label ONCO-TAIN®)
NDC 0009-5093-10
Rx only
Ellence®
(epirubicin hydrochloride injection)
200 mg/100 mL
(2 mg/mL)
For Intravenous Use Only
Single-dose Vial.
Discard unused portion.
Principal Display Panel 200 Mg/100 Ml Vial Carton Onco Tain® (PRINCIPAL DISPLAY PANEL - 200 mg/100 mL Vial Carton ONCO-TAIN®)
NDC 0009-5093-10
One 100 mL Single-dose Vial.
Discard unused portion.
Ellence®
(epirubicin hydrochloride
injection)
200 mg/100 mL
(2 mg/mL)
For Intravenous Use Only
Pfizer
Hospital
Rx only
Warning: Hazardous Drug
5.8 Immunosuppressant Effects/increased Susceptibility to Infections (5.8 Immunosuppressant Effects/Increased Susceptibility to Infections)
Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including epirubicin, may result in serious or fatal infections. Avoid vaccination with a live vaccine in patients receiving ELLENCE. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
Warning: Cardiac Toxicity, Secondary Malignancies, Extravasation and Tissue Necrosis, and Severe Myelosuppression (WARNING: CARDIAC TOXICITY, SECONDARY MALIGNANCIES, EXTRAVASATION AND TISSUE NECROSIS, and SEVERE MYELOSUPPRESSION)
-
•Cardiac Toxicity: Myocardial damage, including acute left ventricular failure, can occur with ELLENCE. The risk of cardiomyopathy is proportional to the cumulative exposure with incidence rates from 0.9% at a cumulative dose of 550 mg/m2, 1.6% at 700 mg/m2, and 3.3% at 900 mg/m2. The risk of cardiomyopathy is further increased with concomitant cardiotoxic therapy. Assess left ventricular ejection fraction (LVEF) before and regularly during and after treatment with ELLENCE [see Warnings and Precautions (5.1)].
-
•Secondary Malignancies: Secondary acute myelogenous leukemia (AML) and myelodysplastic syndrome (MDS) occur at a higher incidence in patients treated with anthracyclines, including ELLENCE [see Warnings and Precautions (5.2)].
-
•Extravasation and Tissue Necrosis: Extravasation of ELLENCE can result in severe local tissue injury and necrosis requiring wide excision of the affected area and skin grafting. Immediately terminate the drug and apply ice to the affected area [see Warnings and Precautions (5.3)].
-
•Severe myelosuppression resulting in serious infection, septic shock, requirement for transfusions, hospitalization, and death may occur [see Warnings and Precautions (5.4)].
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:48:31.151178 · Updated: 2026-03-14T22:30:10.662825