Biktarvy
0476d7eb-1024-4821-bbe7-abaacfda32a2
34391-3
HUMAN PRESCRIPTION DRUG LABEL
Drug Facts
Composition & Product
Identifiers & Packaging
Indications and Usage
BIKTARVY is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 14 kg: who have no antiretroviral treatment history or to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no known or suspected substitutions associated with resistance to bictegravir or tenofovir.
Dosage and Administration
Testing: Prior to or when initiating BIKTARVY test for hepatitis B virus infection. Prior to or when initiating BIKTARVY, and during treatment, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus. ( 2.1 ) Recommended dosage in adults and pediatric patients weighing at least 25 kg: One tablet containing 50 mg BIC, 200 mg FTC, and 25 mg TAF taken once daily with or without food. ( 2.2 ) Recommended dosage in pediatric patients weighing at least 14 kg to less than 25 kg: One tablet containing 30 mg BIC, 120 mg FTC, and 15 mg TAF taken once daily with or without food. ( 2.3 ) Renal impairment: BIKTARVY is not recommended in patients with estimated creatinine clearance of 15 to below 30 mL/min, or below 15 mL/min who are not receiving chronic hemodialysis, or below 15 mL/min who have no antiretroviral treatment history. ( 2.4 ) Hepatic impairment: BIKTARVY is not recommended in patients with severe hepatic impairment. ( 2.5 )
Contraindications
BIKTARVY is contraindicated to be co-administered with: dofetilide due to the potential for increased dofetilide plasma concentrations and associated serious and/or life-threatening events [see Drug Interactions (7.5) ] . rifampin due to decreased BIC plasma concentrations, which may result in the loss of therapeutic effect and development of resistance to BIKTARVY [see Drug Interactions (7.5) ] .
Warnings and Precautions
Immune reconstitution syndrome: May necessitate further evaluation and treatment. ( 5.3 ) New onset or worsening renal impairment: Assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein when initiating BIKTARVY and during therapy as clinically appropriate in all patients. Also assess serum phosphorus in patients with chronic kidney disease. ( 5.4 ) Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. ( 5.5 )
Adverse Reactions
The following adverse reactions are discussed in other sections of the labeling: Severe Acute Exacerbations of Hepatitis B [see Warnings and Precautions (5.1) ] . Immune Reconstitution Syndrome [see Warnings and Precautions (5.3) ] . New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.4) ]. Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.5) ].
Drug Interactions
Table 3 provides a listing of established or potentially clinically significant drug interactions with recommended prevention or management strategies. The drug interactions described are based on studies conducted with either BIKTARVY, the components of BIKTARVY (BIC, FTC, and TAF) as individual agents, or are drug interactions that may occur with BIKTARVY [see Contraindications (4) , Warnings and Precautions (5.2) , and Clinical Pharmacology (12.3) ] . Table 3 Established and Potentially Significant Table is not all inclusive. Drug Interactions: Alteration in Regimen May be Recommended Concomitant Drug Class: Drug Name Effect on Concentration ↑ = Increase, ↓ = Decrease. Clinical Comment Antiarrhythmics: dofetilide ↑ Dofetilide Coadministration is contraindicated due to the potential for serious and/or life-threatening events associated with dofetilide therapy [see Contraindications (4) ] . Anticonvulsants: carbamazepine Drug-drug interaction study was conducted with either BIKTARVY or its components as individual agents. oxcarbazepine phenobarbital phenytoin ↓ BIC ↓ TAF Coadministration with alternative anticonvulsants should be considered. Antimycobacterials: rifabutin rifampin , Strong inducer of CYP3Aand P-gp, and inducer of UGT1A1. rifapentine ↓ BIC ↓ TAF Coadministration with rifampin is contraindicated due to the effect of rifampin on the BIC component of BIKTARVY [see Contraindications (4) ] . Coadministration with rifabutin or rifapentine is not recommended. Herbal Products: St. John's wort The induction potency of St. John's wort may vary widely based on preparation. ↓ BIC ↓ TAF Coadministration with St. John's wort is not recommended. Medications or oral supplements containing polyvalent cations (e.g., Mg, Al, Ca, Fe): Calcium or iron supplements Cation-containing antacids or laxatives Sucralfate Buffered medications ↓ BIC Antacids containing Al/Mg: BIKTARVY can be taken at least 2 hours before or 6 hours after taking antacids containing Al/Mg. Routine administration of BIKTARVY together with, or 2 hours after, antacids containing Al/Mg is not recommended. Supplements or Antacids containing Calcium or Iron: BIKTARVY and supplements or antacids containing calcium or iron can be taken together with food. Routine administration of BIKTARVY under fasting conditions together with, or 2 hours after, supplements or antacids containing calcium or iron is not recommended. Metformin ↑ Metformin Refer to the prescribing information of metformin for assessing the benefit and risk of concomitant use of BIKTARVY and metformin.
How Supplied
Product: 50090-6247 NDC: 50090-6247-0 30 TABLET in a BOTTLE, PLASTIC
Storage and Handling
Product: 50090-6247 NDC: 50090-6247-0 30 TABLET in a BOTTLE, PLASTIC
Description
Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY. Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue BIKTARVY. If appropriate, anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1) ] .
Medication Information
Warnings and Precautions
Immune reconstitution syndrome: May necessitate further evaluation and treatment. ( 5.3 ) New onset or worsening renal impairment: Assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein when initiating BIKTARVY and during therapy as clinically appropriate in all patients. Also assess serum phosphorus in patients with chronic kidney disease. ( 5.4 ) Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. ( 5.5 )
Indications and Usage
BIKTARVY is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 14 kg: who have no antiretroviral treatment history or to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no known or suspected substitutions associated with resistance to bictegravir or tenofovir.
Dosage and Administration
Testing: Prior to or when initiating BIKTARVY test for hepatitis B virus infection. Prior to or when initiating BIKTARVY, and during treatment, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus. ( 2.1 ) Recommended dosage in adults and pediatric patients weighing at least 25 kg: One tablet containing 50 mg BIC, 200 mg FTC, and 25 mg TAF taken once daily with or without food. ( 2.2 ) Recommended dosage in pediatric patients weighing at least 14 kg to less than 25 kg: One tablet containing 30 mg BIC, 120 mg FTC, and 15 mg TAF taken once daily with or without food. ( 2.3 ) Renal impairment: BIKTARVY is not recommended in patients with estimated creatinine clearance of 15 to below 30 mL/min, or below 15 mL/min who are not receiving chronic hemodialysis, or below 15 mL/min who have no antiretroviral treatment history. ( 2.4 ) Hepatic impairment: BIKTARVY is not recommended in patients with severe hepatic impairment. ( 2.5 )
Contraindications
BIKTARVY is contraindicated to be co-administered with: dofetilide due to the potential for increased dofetilide plasma concentrations and associated serious and/or life-threatening events [see Drug Interactions (7.5) ] . rifampin due to decreased BIC plasma concentrations, which may result in the loss of therapeutic effect and development of resistance to BIKTARVY [see Drug Interactions (7.5) ] .
Adverse Reactions
The following adverse reactions are discussed in other sections of the labeling: Severe Acute Exacerbations of Hepatitis B [see Warnings and Precautions (5.1) ] . Immune Reconstitution Syndrome [see Warnings and Precautions (5.3) ] . New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.4) ]. Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.5) ].
Drug Interactions
Table 3 provides a listing of established or potentially clinically significant drug interactions with recommended prevention or management strategies. The drug interactions described are based on studies conducted with either BIKTARVY, the components of BIKTARVY (BIC, FTC, and TAF) as individual agents, or are drug interactions that may occur with BIKTARVY [see Contraindications (4) , Warnings and Precautions (5.2) , and Clinical Pharmacology (12.3) ] . Table 3 Established and Potentially Significant Table is not all inclusive. Drug Interactions: Alteration in Regimen May be Recommended Concomitant Drug Class: Drug Name Effect on Concentration ↑ = Increase, ↓ = Decrease. Clinical Comment Antiarrhythmics: dofetilide ↑ Dofetilide Coadministration is contraindicated due to the potential for serious and/or life-threatening events associated with dofetilide therapy [see Contraindications (4) ] . Anticonvulsants: carbamazepine Drug-drug interaction study was conducted with either BIKTARVY or its components as individual agents. oxcarbazepine phenobarbital phenytoin ↓ BIC ↓ TAF Coadministration with alternative anticonvulsants should be considered. Antimycobacterials: rifabutin rifampin , Strong inducer of CYP3Aand P-gp, and inducer of UGT1A1. rifapentine ↓ BIC ↓ TAF Coadministration with rifampin is contraindicated due to the effect of rifampin on the BIC component of BIKTARVY [see Contraindications (4) ] . Coadministration with rifabutin or rifapentine is not recommended. Herbal Products: St. John's wort The induction potency of St. John's wort may vary widely based on preparation. ↓ BIC ↓ TAF Coadministration with St. John's wort is not recommended. Medications or oral supplements containing polyvalent cations (e.g., Mg, Al, Ca, Fe): Calcium or iron supplements Cation-containing antacids or laxatives Sucralfate Buffered medications ↓ BIC Antacids containing Al/Mg: BIKTARVY can be taken at least 2 hours before or 6 hours after taking antacids containing Al/Mg. Routine administration of BIKTARVY together with, or 2 hours after, antacids containing Al/Mg is not recommended. Supplements or Antacids containing Calcium or Iron: BIKTARVY and supplements or antacids containing calcium or iron can be taken together with food. Routine administration of BIKTARVY under fasting conditions together with, or 2 hours after, supplements or antacids containing calcium or iron is not recommended. Metformin ↑ Metformin Refer to the prescribing information of metformin for assessing the benefit and risk of concomitant use of BIKTARVY and metformin.
Storage and Handling
Product: 50090-6247 NDC: 50090-6247-0 30 TABLET in a BOTTLE, PLASTIC
How Supplied
Product: 50090-6247 NDC: 50090-6247-0 30 TABLET in a BOTTLE, PLASTIC
Description
Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY. Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue BIKTARVY. If appropriate, anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1) ] .
Section 42229-5
Clinical Trials in Adults with No Antiretroviral Treatment History
The primary safety assessment of BIKTARVY was based on data from two randomized, double-blind, active-controlled trials, Trial 1489 and Trial 1490, that enrolled 1274 HIV-1 infected adult subjects with no antiretroviral treatment history through Week 144. After Week 144, subjects received open-label BIKTARVY in an optional extension phase for an additional 96 weeks (end of study). A total of 634 and 1025 subjects received one tablet of BIKTARVY once daily during the double-blind (Week 144) and extension phases, respectively [see Clinical Studies (14.2)].
The most common adverse reactions (all Grades) reported in at least 5% of subjects in the BIKTARVY group in either Trial 1489 or Trial 1490 were diarrhea, nausea, and headache. The proportion of subjects who discontinued treatment through Week 144 with BIKTARVY, abacavir [ABC]/dolutegravir [DTG]/ lamivudine [3TC]), or DTG + FTC/TAF, due to adverse events, regardless of severity, was 1%, 2%, and 2%, respectively. Table 1 displays the frequency of adverse reactions (all Grades) greater than or equal to 2% in the BIKTARVY group.
| Trial 1489 | Trial 1490 | |||
|---|---|---|---|---|
| Adverse Reactions | BIKTARVY N=314 |
ABC/DTG/3TC N=315 |
BIKTARVY N=320 |
DTG + FTC/TAF N=325 |
| Diarrhea | 6% | 4% | 3% | 3% |
| Nausea | 6% | 18% | 3% | 5% |
| Headache | 5% | 5% | 4% | 3% |
| Fatigue | 3% | 4% | 2% | 2% |
| Abnormal dreams | 3% | 3% | <1% | 1% |
| Dizziness | 2% | 3% | 2% | 1% |
| Insomnia | 2% | 3% | 2% | <1% |
| Abdominal distention | 2% | 2% | 1% | 2% |
Additional adverse reactions (all Grades) occurring in less than 2% of subjects administered BIKTARVY in Trials 1489 and 1490 included vomiting, flatulence, dyspepsia, abdominal pain, rash, and depression.
Suicidal ideation, suicide attempt, and depression suicidal occurred in 2% of subjects administered BIKTARVY; these events occurred primarily in subjects with a preexisting history of depression, prior suicide attempt or psychiatric illness.
The majority (84%) of adverse events associated with BIKTARVY were Grade 1.
Adverse reactions in the open-label extension phases of Trials 1489 and 1490 were similar to those observed in subjects administered BIKTARVY in the Week 144 analysis.
Section 42230-3
| Patient Information BIKTARVY® (bik-TAR-vee) (bictegravir, emtricitabine, and tenofovir alafenamide) tablets |
|
|---|---|
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised:02/2024 |
| Important: Ask your healthcare provider or pharmacist about medicines that should not be taken with BIKTARVY. For more information, see "What should I tell my healthcare provider before taking BIKTARVY?" | |
|
What is the most important information I should know about BIKTARVY? |
|
| BIKTARVY can cause serious side effects, including: | |
|
|
| For more information about side effects, see "What are the possible side effects of BIKTARVY?" | |
|
What is BIKTARVY?
BIKTARVY is a prescription medicine that is used without other human immunodeficiency virus-1 (HIV-1) medicines to treat HIV-1 infection in adults and children who weigh at least 31 pounds (14 kg): |
|
|
|
| HIV-1 is the virus that causes Acquired Immune Deficiency Syndrome (AIDS). BIKTARVY contains the medicines bictegravir, emtricitabine, and tenofovir alafenamide. It is not known if BIKTARVY is safe and effective in children who weigh less than 31 pounds (14 kg). |
|
| Do not take BIKTARVY if you also take a medicine that contains: | |
|
|
|
What should I tell my healthcare provider before taking BIKTARVY? |
|
Some medicines may interact with BIKTARVY. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
|
|
How should I take BIKTARVY?
|
|
|
What are the possible side effects of BIKTARVY? |
|
| BIKTARVY may cause serious side effects, including: | |
|
|
| The most common side effects of BIKTARVY are diarrhea, nausea, and headache. | |
| These are not all of the possible side effects of BIKTARVY. | |
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |
How should I store BIKTARVY?
|
|
| Keep BIKTARVY and all medicines out of reach of children. | |
| General information about the safe and effective use of BIKTARVY. | |
| Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use BIKTARVY for a condition for which it was not prescribed. Do not give BIKTARVY to other people, even if they have the same symptoms you have. It may harm them. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about BIKTARVY that is written for health professionals. | |
| What are the ingredients in BIKTARVY? | |
| Active ingredients: bictegravir, emtricitabine, and tenofovir alafenamide. | |
| Inactive ingredients: croscarmellose sodium, magnesium stearate, and microcrystalline cellulose. | |
| The tablets are film-coated with a coating material containing iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. | |
| Manufactured and distributed by: Gilead Sciences, Inc. Foster City, CA 94404 | |
| BIKTARVY is a trademark of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners. | |
| © 2024 Gilead Sciences, Inc. All rights reserved. 210251-GS-012 | |
| For more information, call 1-800-445-3235 or go to www.BIKTARVY.com. |
Section 43683-2
| Indications and Usage (1) | 02/2024 |
Biktarvy
10 Overdosage
No data are available on overdose of BIKTARVY in patients. If overdose occurs, monitor the patient for evidence of toxicity. Treatment of overdose with BIKTARVY consists of general supportive measures including monitoring of vital signs as well as observation of the clinical status of the patient.
Hemodialysis treatment removes approximately 30% of the FTC dose over a 3-hour dialysis period starting within 1.5 hours of FTC dosing (blood flow rate of 400 mL/min and a dialysate flow rate of 600 mL/min). It is not known whether FTC can be removed by peritoneal dialysis.
Tenofovir is efficiently removed by hemodialysis with an extraction coefficient of approximately 54%.
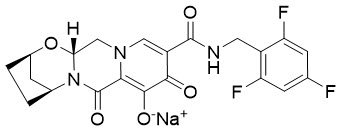
11 Description
BIKTARVY (bictegravir, emtricitabine, and tenofovir alafenamide) is a fixed dose combination tablet containing bictegravir (BIC), emtricitabine (FTC), and tenofovir alafenamide (TAF) for oral administration.
- BIC is an integrase strand transfer inhibitor (INSTI).
- FTC, a synthetic nucleoside analog of cytidine, is an HIV nucleoside analog reverse transcriptase inhibitor (HIV NRTI).
- TAF, an HIV NRTI, is converted in vivo to tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate.
BIKTARVY tablets are available in two dose strengths:
- 50 mg/200 mg/25 mg tablet containing 50 mg of BIC (equivalent to 52.5 mg of bictegravir sodium),
200 mg of FTC, and 25 mg of TAF (equivalent to 28 mg of tenofovir alafenamide fumarate). - 30 mg/120 mg/15 mg tablet containing 30 mg of BIC (equivalent to 31.5 mg of bictegravir sodium), 120 mg of FTC, and 15 mg of TAF (equivalent to 16.8 mg of tenofovir alafenamide fumarate).
Both dose strengths of BIKTARVY tablets include the following inactive ingredients: croscarmellose sodium, magnesium stearate, and microcrystalline cellulose. The tablets for both dose strengths are film-coated with a coating material containing iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
8.4 Pediatric Use
The safety and effectiveness of BIKTARVY have been established as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in pediatric patients weighing at least 14 kg:
- who have no antiretroviral treatment history or
- to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no known or suspected resistance to bictegravir or tenofovir [see Indications and Usage (1), and Dosage and Administration (2.2, 2.3)].
Use of BIKTARVY in pediatric patients weighing at least 14 kg is supported by the following:
- trials in adults [see Clinical Studies (14.1)]
- an open-label trial in three age-based cohorts of virologically-suppressed pediatric subjects [see Clinical Studies (14.4)]
- Cohort 1: 12 to less than 18 years of age and weighing at least 35 kg receiving BIKTARVY through Week 48 (N=50),
- Cohort 2: 6 to less than 12 years of age and weighing at least 25 kg receiving BIKTARVY through Week 24 (N=50), and
- Cohort 3: at least 2 years of age and weighing at least 14 to less than 25 kg through Week 24 (N=22). No pediatric subjects 2 years of age were enrolled; of the 6 pediatric subjects who were 3 years of age at enrollment, 3 subjects weighed between 14 to less than 15 kg.
The safety and efficacy of BIKTARVY in these pediatric subjects were similar to that in adults, and there was no clinically significant change in exposure for the components of BIKTARVY [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.4)].
Safety and effectiveness of BIKTARVY in pediatric patients weighing less than 14 kg have not been established.
8.5 Geriatric Use
Clinical trials in virologically-suppressed subjects (Trials 4449, 1844, and 1878) included 111 subjects aged 65 years and over who received BIKTARVY, including 86 patients from an open-label, single-arm trial of subjects aged 65 years and over who were switched from their previous antiretroviral regimen to BIKTARVY [see Clinical Studies (14.3) ]. Of the total number of BIKTARVY-treated patients in these trials, 100 (90%) were 65 to 74 years of age, and 11 (10%) were 75 to 84 years of age. No overall differences in safety or effectiveness were observed between elderly subjects and adults between 18 and less than 65 years of age, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
4 Contraindications
BIKTARVY is contraindicated to be co-administered with:
- dofetilide due to the potential for increased dofetilide plasma concentrations and associated serious and/or life-threatening events [see Drug Interactions (7.5)].
- rifampin due to decreased BIC plasma concentrations, which may result in the loss of therapeutic effect and development of resistance to BIKTARVY [see Drug Interactions (7.5)].
6 Adverse Reactions
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbations of Hepatitis B [see Warnings and Precautions (5.1)].
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.3)].
- New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.4)].
- Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.5)].
7 Drug Interactions
8.6 Renal Impairment
The pharmacokinetics, safety, virologic and immunologic responses of FTC and TAF (components of BIKTARVY) were evaluated in a single arm, open-label trial (Trial 1825) in virologically-suppressed adults with ESRD (estimated creatinine clearance of less than 15 mL/min) on chronic hemodialysis treated with FTC+TAF in combination with elvitegravir and cobicistat as a fixed-dose combination tablet for 96 weeks (N=55). In an extension phase of Trial 1825, 10 virologically-suppressed subjects switched to BIKTARVY and all remained virologically suppressed for 48 weeks [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.3)].
No dosage adjustment of BIKTARVY is recommended in patients with estimated creatinine clearance greater than or equal to 30 mL/min, or in virologically-suppressed adults (estimated creatinine clearance below 15 mL/min) who are receiving chronic hemodialysis. On days of hemodialysis, administer the daily dose of BIKTARVY after completion of hemodialysis treatment [see Dosage and Administration (2.2)]
BIKTARVY is not recommended in patients with estimated creatinine clearance of below 30 mL/min, by Cockcroft-Gault, or patients with ESRD (estimated creatinine clearance below 15 mL/min) who are not receiving chronic dialysis, or patients with no antiretroviral treatment history and ESRD who are receiving chronic dialysis, as the safety and/or efficacy of BIKTARVY has not been established in these populations [see Dosage and Administration (2.4), Warnings and Precautions (5.4), and Clinical Pharmacology (12.3)].
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of BIKTARVY components are provided in Table 4. The multiple dose PK parameters of BIKTARVY components (based on population pharmacokinetic analysis) are provided in Table 5.
| Bictegravir (BIC) | Emtricitabine (FTC) | Tenofovir Alafenamide (TAF) | ||
|---|---|---|---|---|
| PBMCs=peripheral blood mononuclear cells; CES1=carboxylesterase 1 | ||||
| Absorption | ||||
| Tmax (h) Values reflect administration of BIKTARVY with or without food.
|
2.0–4.0 | 1.5–2.0 | 0.5–2.0 | |
| Effect of high-fat meal (relative to fasting) Values refer to geometric mean ratio [high-fat meal/ fasting] in PK parameters and (90% confidence interval). High fat meal is approximately 800 kcal, 50% fat.
|
AUC ratio | 1.24 (1.16, 1.33) | 0.96 (0.93, 0.99) | 1.63 (1.43, 1.85) |
| Cmax ratio | 1.13 (1.06, 1.20) | 0.86 (0.78, 0.93) | 0.92 (0.73, 1.14) | |
| Distribution | ||||
| % bound to human plasma proteins | >99 | <4 | ~80 | |
| Blood-to-plasma ratio | 0.64 | 0.6 | 1.0 | |
| Elimination | ||||
| t1/2 (h) t1/2 values refer to median (Q1, Q3) terminal plasma half-life. Note that the active metabolite of TAF, tenofovir diphosphate, has a half-life of 150–180 hours within PBMCs.
|
17.3 (14.8, 20.7) | 10.4 (9.0, 12.0) | 0.51 (0.45, 0.62) | |
| Metabolism | ||||
| Metabolic pathway(s) | CYP3A UGT1A1 |
Not significantly metabolized | Cathepsin A
In vivo, TAF is hydrolyzed within cells to form tenofovir (major metabolite), which is phosphorylated to the active metabolite, tenofovir diphosphate. In vitro studies have shown that TAF is metabolized to tenofovir by cathepsin A in PBMCs and macrophages; and by CES1 in hepatocytes. (PBMCs)CES1 (hepatocytes) |
|
| Excretion | ||||
| Major route of elimination | Metabolism | Glomerular filtration and active tubular secretion | Metabolism | |
| % of dose excreted in urine Dosing in mass balance studies: single dose administration of [14C] BIC; single dose administration of [14C] FTC after multiple dosing of FTC for ten days; single dose administration of [14C] TAF.
|
35 | 70 | <1 | |
| % of dose excreted in feces | 60.3 | 13.7 | 31.7 |
| Parameter Mean (CV%) | Bictegravir | Emtricitabine | Tenofovir Alafenamide |
|---|---|---|---|
| CV=Coefficient of Variation; NA=Not Applicable | |||
| Cmax
(microgram per mL) |
6.15 (22.9) | 2.13 (34.7) | 0.121 (15.4) |
| AUCtau
(microgram∙h per mL) |
102 (26.9) | 12.3 (29.2) | 0.142 (17.3) |
| Ctrough
(microgram per mL) |
2.61 (35.2) | 0.096 (37.4) | NA |
8.7 Hepatic Impairment
No dosage adjustment of BIKTARVY is recommended in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. BIKTARVY has not been studied in patients with severe hepatic impairment (Child-Pugh Class C). Therefore, BIKTARVY is not recommended for use in patients with severe hepatic impairment [see Dosage and Administration (2.4), and Clinical Pharmacology (12.3)].
1 Indications and Usage
BIKTARVY is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 14 kg:
- who have no antiretroviral treatment history or
- to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no known or suspected substitutions associated with resistance to bictegravir or tenofovir.
12.1 Mechanism of Action
BIKTARVY is a fixed dose combination of antiretroviral drugs bictegravir (BIC), emtricitabine (FTC), and tenofovir alafenamide (TAF) [see Microbiology (12.4)].
5 Warnings and Precautions
- Immune reconstitution syndrome: May necessitate further evaluation and treatment. (5.3)
- New onset or worsening renal impairment: Assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein when initiating BIKTARVY and during therapy as clinically appropriate in all patients. Also assess serum phosphorus in patients with chronic kidney disease. (5.4)
- Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. (5.5)
2 Dosage and Administration
- Testing: Prior to or when initiating BIKTARVY test for hepatitis B virus infection. Prior to or when initiating BIKTARVY, and during treatment, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus. (2.1)
- Recommended dosage in adults and pediatric patients weighing at least 25 kg: One tablet containing 50 mg BIC, 200 mg FTC, and 25 mg TAF taken once daily with or without food. (2.2)
- Recommended dosage in pediatric patients weighing at least 14 kg to less than 25 kg: One tablet containing 30 mg BIC, 120 mg FTC, and 15 mg TAF taken once daily with or without food. (2.3)
- Renal impairment: BIKTARVY is not recommended in patients with estimated creatinine clearance of 15 to below 30 mL/min, or below 15 mL/min who are not receiving chronic hemodialysis, or below 15 mL/min who have no antiretroviral treatment history. (2.4)
- Hepatic impairment: BIKTARVY is not recommended in patients with severe hepatic impairment. (2.5)
3 Dosage Forms and Strengths
BIKTARVY tablets are available in two dose strengths:
- 50 mg/200 mg/25 mg tablets: 50 mg of bictegravir (BIC) (equivalent to 52.5 mg of bictegravir sodium), 200 mg of emtricitabine (FTC), and 25 mg of tenofovir alafenamide (TAF) (equivalent to 28 mg of tenofovir alafenamide fumarate). These tablets are purplish brown, capsule-shaped, film-coated, and debossed with "GSI" on one side and "9883" on the other side.
- 30 mg/120 mg/15 mg tablets: 30 mg of BIC (equivalent to 31.5 mg of bictegravir sodium), 120 mg of FTC, and 15 mg of TAF (equivalent to 16.8 mg of tenofovir alafenamide fumarate). These tablets are pink, capsule-shaped, film-coated, and debossed with "GSI" on one side and "B" on the other side.
6.2 Postmarketing Experience
The following events have been identified during post approval use of BIKTARVY or products containing TAF. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
8 Use in Specific Populations
- Pediatrics: Not recommended for patients weighing less than 14 kg. (8.4)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
5.3 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections [such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia (PCP), or tuberculosis], which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
7.4 Drugs Affecting Renal Function
Because FTC and tenofovir are primarily excreted by the kidneys by a combination of glomerular filtration and active tubular secretion, coadministration of BIKTARVY with drugs that reduce renal function or compete for active tubular secretion may increase concentrations of FTC, tenofovir, and other renally eliminated drugs and this may increase the risk of adverse reactions. Some examples of drugs that are eliminated by active tubular secretion include, but are not limited to, acyclovir, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.4)].
14.1 Description of Clinical Trials
The efficacy and safety of BIKTARVY were evaluated in the trials summarized in Table 12.
| Trial | Population | Trial Arms (N) | Timepoint (Week) |
|---|---|---|---|
| OLE = open-label extension | |||
| Trial 1489 Randomized, double blind, active controlled trial.
(NCT 02607930) |
Adults with no antiretroviral treatment history | BIKTARVY (314) ABC/DTG/3TC (315) |
144 + 96 (OLE) 144-week double-blind active controlled phase followed by an extension phase in which 1025 subjects from Trials 1489 and 1490 received open-label BIKTARVY for 96 weeks.
|
| Trial 1490
(NCT 02607956) |
BIKTARVY (320) DTG + FTC/TAF(325) |
144 + 96 (OLE) | |
| Trial 1844
(NCT 02603120) |
Virologically-suppressed HIV-1 RNA less than 50 copies per mL. adults |
BIKTARVY (282) ABC/DTG/3TC (281) |
48 |
| Trial 1878 Randomized, open-label, active controlled trial.
(NCT 02603107) |
BIKTARVY (290) ATV or DRV (with cobicistat or ritonavir) plus either FTC/TDF or ABC/3TC (287) |
48 | |
| Trial 4030
(NCT 03110380) |
BIKTARVY (284 [47 with M184V/I]) DTG plus FTC/TAF (281 [34 with M184V/I]) |
48 | |
| Trial 1825 Open-label trial.
(NCT 02600819) |
Virologically-suppressed adults with ESRD End stage renal disease (estimated creatinine clearance of less than 15 mL/min by Cockcroft-Gault method). receiving chronic hemodialysis |
FTC+TAF in combination with elvitegravir and cobicistat as a fixed-dose combination (55). In an extension phase of Trial 1825, 10 virologically-suppressed subjects switched to BIKTARVY. | 48 Subjects received FTC+TAF in combination with elvitegravir and cobicistat for 96 weeks, followed by an extension phase in which 10 subjects received BIKTARVY for 48 weeks.
|
| Trial 4449
(NCT 03405935) |
Virologically-suppressed adults aged 65 years and over | BIKTARVY (86) | 48 |
| Trial 1474
(cohort 1) (NCT 02881320) |
Virologically-suppressed adolescents between the ages of 12 to less than 18 years (at least 35 kg) | BIKTARVY (50) | 48 |
| Trial 1474
(cohort 2) (NCT 02881320) |
Virologically-suppressed children between the ages of 6 to less than 12 years (at least 25 kg) | BIKTARVY (50) | 24 |
| Trial 1474
(cohort 3) (NCT 02881320) |
Virologically-suppressed children at least 2 years of age (at least 14 to less than 25 kg) | BIKTARVY (22) | 24 |
16 How Supplied/storage and Handling
Product: 50090-6247
NDC: 50090-6247-0 30 TABLET in a BOTTLE, PLASTIC
7.1 Other Antiretroviral Medications
Because BIKTARVY is a complete regimen, coadministration with other antiretroviral medications for the treatment of HIV-1 infection is not recommended [see Indications and Usage (1)]. Comprehensive information regarding potential drug-drug interactions with other antiretroviral medications is not provided because the safety and efficacy of concomitant HIV-1 antiretroviral therapy is unknown.
13.2 Animal Toxicology And/or Pharmacology
Minimal to slight infiltration of mononuclear cells in the posterior uvea was observed in dogs with similar severity after three and nine month administration of TAF; reversibility was seen after a three month recovery period. No eye toxicity was observed in the dog at systemic exposures of 7 (TAF) and 14 (tenofovir) times the exposure seen in humans with the recommended daily dose of BIKTARVY.
5.4 New Onset Or Worsening Renal Impairment
Postmarketing cases of renal impairment, including acute renal failure, proximal renal tubulopathy (PRT), and Fanconi syndrome, have been reported with TAF-containing products; while most of these cases were characterized by potential confounders that may have contributed to the reported renal events, it is also possible these factors may have predisposed patients to tenofovir-related adverse events [see Adverse Reactions (6.1, 6.2)]. BIKTARVY is not recommended in patients with severe renal impairment (estimated creatinine clearance of 15 to below 30 mL/min), or patients with ESRD (estimated creatinine clearance below 15 mL/min) who are not receiving chronic hemodialysis, or patients with no antiretroviral treatment history and ESRD who are receiving chronic hemodialysis [see Dosage and Administration (2.4), and Use in Specific Populations (8.6)].
Patients taking tenofovir prodrugs who have impaired renal function and those taking nephrotoxic agents including non-steroidal anti-inflammatory drugs are at increased risk of developing renal-related adverse reactions.
Prior to or when initiating BIKTARVY, and during treatment with BIKTARVY, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus. Discontinue BIKTARVY in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome.
7.2 Potential for Biktarvy to Affect Other Drugs
BIC inhibits organic cation transporter 2 (OCT2) and multidrug and toxin extrusion transporter 1 (MATE1) in vitro. Coadministration of BIKTARVY with drugs that are substrates of OCT2 and MATE1 (e.g., dofetilide) may increase their plasma concentrations (see Table 3).
5.5 Lactic Acidosis/severe Hepatomegaly With Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including emtricitabine, a component of BIKTARVY, and tenofovir DF, another prodrug of tenofovir, alone or in combination with other antiretrovirals. Treatment with BIKTARVY should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
Warning: Post Treatment Acute Exacerbation of Hepatitis B
Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY.
Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue BIKTARVY. If appropriate, anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)].
14.4 Clinical Trial Results in Pediatric Subjects With Hiv 1
In Trial 1474, an open-label, single arm trial the efficacy, safety, and pharmacokinetics of BIKTARVY in HIV-1 infected pediatric subjects were evaluated in virologically-suppressed adolescents between the ages of 12 to less than 18 years weighing at least 35 kg (N=50), in virologically-suppressed children between the ages of 6 to less than 12 years weighing at least 25 kg (N=50), and in virologically-suppressed children at least 2 years of age and weighing at least 14 to less than 25 kg (N=22).
2.4 Not Recommended in Patients With Severe Renal Impairment
BIKTARVY is not recommended in patients with [see Dosage and Administration (2.2, 2.3), and Use in Specific Populations (8.6)]:
- severe renal impairment (estimated creatinine clearance of 15 to below 30 mL/min); or
- end stage renal disease (ESRD; estimated creatinine clearance below 15 mL/min who are not receiving chronic hemodialysis; or
- no antiretroviral treatment history and ESRD who are receiving chronic hemodialysis.
7.5 Established and Potentially Significant Drug Interactions
Table 3 provides a listing of established or potentially clinically significant drug interactions with recommended prevention or management strategies. The drug interactions described are based on studies conducted with either BIKTARVY, the components of BIKTARVY (BIC, FTC, and TAF) as individual agents, or are drug interactions that may occur with BIKTARVY [see Contraindications (4), Warnings and Precautions (5.2), and Clinical Pharmacology (12.3)].
| Concomitant Drug Class: Drug Name | Effect on Concentration ↑ = Increase, ↓ = Decrease.
|
Clinical Comment |
|---|---|---|
|
Antiarrhythmics:
dofetilide |
↑ Dofetilide | Coadministration is contraindicated due to the potential for serious and/or life-threatening events associated with dofetilide therapy [see Contraindications (4)]. |
|
Anticonvulsants:
carbamazepine Drug-drug interaction study was conducted with either BIKTARVY or its components as individual agents.
oxcarbazepine phenobarbital phenytoin |
↓ BIC ↓ TAF |
Coadministration with alternative anticonvulsants should be considered. |
|
Antimycobacterials:
rifabutin rifampin , Strong inducer of CYP3Aand P-gp, and inducer of UGT1A1.
rifapentine |
↓ BIC ↓ TAF |
Coadministration with rifampin is contraindicated due to the effect of rifampin on the BIC component of BIKTARVY [see Contraindications (4)]. Coadministration with rifabutin or rifapentine is not recommended. |
|
Herbal Products:
St. John's wort The induction potency of St. John's wort may vary widely based on preparation.
|
↓ BIC ↓ TAF |
Coadministration with St. John's wort is not recommended. |
|
Medications or oral supplements containing polyvalent cations (e.g., Mg, Al, Ca, Fe):
Calcium or iron supplements Cation-containing antacids or laxatives Sucralfate Buffered medications |
↓ BIC |
Antacids containing Al/Mg:
BIKTARVY can be taken at least 2 hours before or 6 hours after taking antacids containing Al/Mg. Routine administration of BIKTARVY together with, or 2 hours after, antacids containing Al/Mg is not recommended. Supplements or Antacids containing Calcium or Iron: BIKTARVY and supplements or antacids containing calcium or iron can be taken together with food. Routine administration of BIKTARVY under fasting conditions together with, or 2 hours after, supplements or antacids containing calcium or iron is not recommended. |
| Metformin | ↑ Metformin | Refer to the prescribing information of metformin for assessing the benefit and risk of concomitant use of BIKTARVY and metformin. |
2.1 Testing When Initiating and During Treatment With Biktarvy
Prior to or when initiating BIKTARVY, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to or when initiating BIKTARVY, and during treatment with BIKTARVY, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus [see Warnings and Precautions (5.4)].
2.5 Not Recommended in Patients With Severe Hepatic Impairment
BIKTARVY is not recommended in patients with severe hepatic impairment (Child-Pugh Class C) [see Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
7.6 Drugs Without Clinically Significant Interactions With Biktarvy
Based on drug interaction studies conducted with BIKTARVY or the components of BIKTARVY, no clinically significant drug interactions have been observed when BIKTARVY is combined with the following drugs: ethinyl estradiol, ledipasvir/sofosbuvir, midazolam, norgestimate, sertraline, sofosbuvir, sofosbuvir/velpatasvir, and sofosbuvir/velpatasvir/voxilaprevir.
7.3 Potential Effect of Other Drugs On One Or More Components of Biktarvy
BIC is a substrate of CYP3A and UGT1A1. A drug that is a strong inducer of CYP3A and also an inducer of UGT1A1 can substantially decrease the plasma concentrations of BIC which may lead to loss of therapeutic effect of BIKTARVY and development of resistance [see Clinical Pharmacology (12.3)].
The use of BIKTARVY with a drug that is a strong inhibitor of CYP3A and also an inhibitor of UGT1A1 may significantly increase the plasma concentrations of BIC.
TAF is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Co-administration of drugs that inhibit P-gp and BCRP may increase the absorption and plasma concentrations of TAF [see Clinical Pharmacology (12.3)]. Co-administration of drugs that induce P-gp activity are expected to decrease the absorption of TAF, resulting in decreased plasma concentration of TAF, which may lead to loss of therapeutic effect of BIKTARVY and development of resistance (see Table 3).
2.2 Recommended Dosage in Adults and Pediatric Patients Weighing At Least 25 Kg
BIKTARVY is a three-drug fixed dose combination product containing bictegravir (BIC), emtricitabine (FTC), and tenofovir alafenamide (TAF). The recommended dosage of BIKTARVY is one tablet containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF taken orally once daily with or without food in:
- adults and pediatric patients weighing at least 25 kg with an estimated creatinine clearance greater than or equal to 30 mL/min; or
- virologically-suppressed adults with an estimated creatinine clearance below 15 mL/min who are receiving chronic hemodialysis. On days of hemodialysis, administer the daily dose of BIKTARVY after completion of hemodialysis treatment [see Use in Specific Populations (8.4, 8.6), and Clinical Pharmacology (12.3)].
5.2 Risk of Adverse Reactions Or Loss of Virologic Response Due to Drug Interactions
The concomitant use of BIKTARVY with certain other drugs may result in known or potentially significant drug interactions, some of which may lead to [see Contraindications (4), and Drug Interactions (7.5)]:
- Loss of therapeutic effect of BIKTARVY and possible development of resistance.
- Possible clinically significant adverse reactions from greater exposures of concomitant drugs.
See Table 3 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during BIKTARVY therapy; review concomitant medications during BIKTARVY therapy; and monitor for the adverse reactions associated with the concomitant drugs.
5.1 Severe Acute Exacerbation of Hepatitis B in Patients Coinfected With Hiv 1 and Hbv
Patients with HIV-1 should be tested for the presence of chronic hepatitis B virus (HBV) infection before or when initiating antiretroviral therapy [see Dosage and Administration (2.1)].
Severe acute exacerbations of hepatitis B (e.g., liver decompensation and liver failure) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing FTC and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY. Patients coinfected with HIV-1 and HBV who discontinue BIKTARVY should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since post-treatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
2.3 Recommended Dosage in Pediatric Patients Weighing At Least 14 Kg to Less Than 25 Kg
The recommended dosage of BIKTARVY is one tablet containing 30 mg of BIC, 120 mg of FTC, and 15 mg of TAF taken orally once daily with or without food in:
- pediatric patients weighing at least 14 kg to less than 25 kg with an estimated creatinine clearance greater than or equal to 30 mL/min [see Use in Specific Populations (8.4, 8.6), and Clinical Pharmacology (12.3)].
For children unable to swallow a whole tablet, the tablet can be split and each part taken separately as long as all parts are ingested within approximately 10 minutes.
14.2 Clinical Trial Results in Adults With Hiv 1 and No Antiretroviral Treatment History
In Trial 1489, adults were randomized in a 1:1 ratio to receive either BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) (N=314) or ABC/DTG/3TC (600 mg/50 mg/300 mg) (N=315) once daily. In Trial 1490, subjects were randomized in a 1:1 ratio to receive either BIKTARVY (N=320) or DTG + FTC/TAF (50 mg + 200 mg/25 mg) (N=325) once daily.
In Trial 1489, the mean age was 34 years (range 18–71), 90% were male, 57% were White, 36% were Black, and 3% were Asian. 22% of patients identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.4 log10 copies/mL (range 1.3–6.5). The mean baseline CD4+ cell count was 464 cells per mm3 (range 0–1424) and 11% had CD4+ cell counts less than 200 cells per mm3. 16% of subjects had baseline viral loads greater than 100,000 copies per mL.
In Trial 1490, the mean age was 37 years (range 18–77), 88% were male, 59% were White, 31% were Black, and 3% were Asian. 25% of patients identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.4 log10 copies/mL (range 2.3–6.6). The mean baseline CD4+ cell count was 456 cells per mm3 (range 2–1636) and 12% had CD4+ cell counts less than 200 cells per mm3. 19% of subjects had baseline viral loads greater than 100,000 copies per mL.
In both trials, subjects were stratified by baseline HIV-1 RNA (less than or equal to 100,000 copies per mL, greater than 100,000 copies per mL to less than or equal to 400,000 copies per mL, or greater than 400,000 copies per mL), by CD4 count (less than 50 cells per mm3, 50–199 cells per mm3, or greater than or equal to 200 cells per mm3), and by region (US or ex-US).
Treatment outcomes of Trials 1489 and 1490 through Week 144 are presented in Table 13.
| Trial 1489 | Trial 1490 | |||
|---|---|---|---|---|
| BIKTARVY (N=314) |
ABC/DTG/3TC (N=315) |
BIKTARVY (N=320) |
DTG + FTC/TAF (N=325) |
|
| HIV-1 RNA < 50 copies/mL | 82% | 84% | 82% | 84% |
| Treatment Difference (95% CI) BIKTARVY vs. Comparator | -2.6% (-8.5% to 3.4%) | -1.9% (-7.8% to 3.9%) | ||
|
HIV-1 RNA ≥ 50 copies/mL Includes subjects who had ≥ 50 copies/mL in the Week 144 window; subjects who discontinued early due to lack or loss of efficacy; subjects who discontinued for reasons other than an adverse event (AE), death or lack or loss of efficacy and at the time of discontinuation had a viral value of ≥ 50 copies/mL.
|
1% | 3% | 5% | 3% |
| No Virologic Data at Week 144 Window | 18% | 13% | 13% | 13% |
| Discontinued Study Drug Due to AE or Death Includes subjects who discontinued due to AE or death at any time point from Day 1 through the time window if this resulted in no virologic data on treatment during the specified window.
|
1% | 2% | 3% | 3% |
| Discontinued Study Drug Due to Other Reasons and Last Available HIV-1 RNA <50 copies/mL Includes subjects who discontinued for reasons other than an AE, death, or lack or loss of efficacy, e.g., withdrew consent, loss to follow-up, etc.
|
16% | 11% | 11% | 9% |
| Missing Data During Window but on Study Drug | 1% | <1% | 0% | 1% |
Treatment outcomes were similar across subgroups by age, sex, race, baseline viral load, and baseline CD4+ cell count.
In Trials 1489 and 1490, the mean increase from baseline in CD4+ count at Week 144 was 299 and 317 cells per mm3 in the BIKTARVY and ABC/DTG/3TC groups, respectively, and 278 and 289 cells per mm3 in the BIKTARVY and DTG + FTC/TAF groups, respectively.
14.3 Clinical Trial Results in Adults With Virologically Suppressed Hiv 1 Who Switched to Biktarvy
In Trial 1844, the efficacy and safety of switching from a regimen of DTG + ABC/3TC or ABC/DTG/3TC to BIKTARVY were evaluated in a randomized, double-blind trial of virologically-suppressed (HIV-1 RNA less than 50 copies per mL) HIV-1 infected adults (N=563, randomized and dosed). Subjects must have been stably suppressed (HIV-1 RNA less than 50 copies per mL) on their baseline regimen for at least 3 months prior to trial entry and had no history of treatment failure. Subjects were randomized in a 1:1 ratio to either switch to BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) at baseline (N=282), or stay on their baseline antiretroviral regimen (N=281). Subjects had a mean age of 45 years (range 20–71), 89% were male, 73% were White, and 22% were Black. 17% of subjects identified as Hispanic/Latino. The mean baseline CD4+ cell count was 723 cells per mm3 (range 124–2444).
In Trial 1878, the efficacy and safety of switching from either ABC/3TC or FTC/TDF (200/300 mg) plus ATV or DRV (given with either cobicistat or ritonavir) to BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) were evaluated in a randomized, open-label study of virologically-suppressed HIV-1 infected adults (N=577, randomized and dosed). Subjects must have been stably suppressed on their baseline regimen for at least 6 months, must not have been previously treated with any INSTI, and had no history of treatment failure. Subjects were randomized in a 1:1 ratio to either switch to BIKTARVY (N=290) or stay on their baseline antiretroviral regimen (N=287). Subjects had a mean age of 46 years (range 20–79), 83% were male, 66% were White, and 26% were Black. 19% of subjects identified as Hispanic/Latino. The mean baseline CD4+ cell count was 663 cells per mm3 (range 62–2582). Subjects were stratified by prior treatment regimen. At screening, 15% of subjects were receiving ABC/3TC plus ATV or DRV (given with either cobicistat or ritonavir) and 85% of subjects were receiving FTC/TDF plus ATV or DRV (given with either cobicistat or ritonavir).
Treatment outcomes of Trials 1844 and 1878 through Week 48 are presented in Table 14.
| Trial 1844 | Trial 1878 | |||
|---|---|---|---|---|
| BIKTARVY (N=282) |
ABC/DTG/3TC (N=281) |
BIKTARVY (N=290) |
ATV- or DRV-based regimen ATV given with cobicistat or ritonavir or DRV given with cobicistat or ritonavir plus either FTC/TDF or ABC/3TC.
(N=287) |
|
|
HIV-1 RNA ≥ 50 copies/mL Includes subjects who had ≥ 50 copies/mL in the Week 48 window; subjects who discontinued early due to lack or loss of efficacy; subjects who discontinued for reasons other than lack or loss of efficacy and at the time of discontinuation had a viral value of ≥ 50 copies/mL.
|
1% | <1% | 2% | 2% |
| Treatment Difference (95% CI) | 0.7% (-1.0% to 2.8%) | 0.0% (-2.5% to 2.5%) | ||
| HIV-1 RNA < 50 copies/mL | 94% | 95% | 92% | 89% |
| No Virologic Data at Week 48 Window | 5% | 5% | 6% | 9% |
| Discontinued Study Drug Due to AE or Death and Last Available HIV-1 RNA < 50 copies/mL | 2% | 1% | 1% | 1% |
| Discontinued Study Drug Due to Other Reasons and Last Available HIV-1 RNA < 50 copies/mL Includes subjects who discontinued for reasons other than an AE, death, or lack or loss of efficacy, e.g., withdrew consent, loss to follow-up, etc.
|
2% | 3% | 3% | 7% |
| Missing Data During Window but on Study Drug | 2% | 1% | 2% | 2% |
In Trial 1844, treatment outcomes between treatment groups were similar across subgroups by age, sex, race, and region. The mean change from baseline in CD4+ count at Week 48 was -31 cells per mm3 in subjects who switched to BIKTARVY and 4 cells per mm3 in subjects who stayed on ABC/DTG/3TC.
In Trial 1878, treatment outcomes between treatment groups were similar across subgroups by age, sex, race, and region. The mean change from baseline in CD4+ count at Week 48 was 25 cells per mm3 in patients who switched to BIKTARVY and 0 cells per mm3 in patients who stayed on their baseline regimen.
In Trial 4030, the efficacy and safety of switching from DTG plus either FTC/TAF or FTC/TDF to BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) were evaluated in a randomized, double-blind study of virologically suppressed HIV-1 infected adults. Subjects must have been stably suppressed (HIV-1 RNA less than 50 copies/mL) on their baseline regimen for at least 6 months (if documented or suspected NRTI resistance), or at least 3 months (if no documented or suspected NRTI resistance) prior to trial entry. Subjects were randomized to switch to BIKTARVY (N=284) or to continue their prior treatment regimen, DTG+ F/TAF (N=281). The primary endpoint was the proportion of subjects with HIV-1 RNA ≥ 50 copies/mL at Week 48. At Week 48 the proportion of subjects with HIV-1 RNA ≥50 copies/mL was 0.4% (1/284) in the BIKTARVY group and 1.1% (3/281) in the DTG+F/TAF group (difference -0.7% [95%CI: -2.8%, 1.0%]).
Of the subjects receiving BIKTARVY, 47 had HIV-1 with pre-existing M184V or I resistance substitutions (M184M/V, M184M/I, M184V/I, M184V) in HIV-1 RT. Eighty-nine percent (42/47) of subjects with M184V or I remained suppressed (HIV-1 RNA < 50 copies/mL) and 11% (5/47 subjects) did not have virologic data at the Week 48 timepoint due to study drug discontinuation.
In Trial 1825, an open-label single arm trial, the efficacy, safety, and pharmacokinetics of FTC and TAF (components of BIKTARVY) were evaluated in virologically-suppressed adults with ESRD (estimated creatinine clearance of less than 15 mL/min) on chronic hemodialysis treated with FTC+TAF in combination with elvitegravir and cobicistat as a fixed-dose combination tablet for 96 weeks (N=55). In an extension phase of Trial 1825, 10 virologically-suppressed subjects switched to BIKTARVY and all subjects remained virologically suppressed (HIV-1 RNA < 50 copies/mL) for 48 weeks.
In Trial 4449, the efficacy and safety of switching from a stable antiretroviral regimen to BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) were evaluated in an open-label, single arm trial of virologically-suppressed (HIV-1 RNA less than 50 copies per mL) HIV-1 infected adults aged 65 years and over (N=86). Subjects treated with BIKTARVY had a mean age of 70 years (range: 65 to 80). The primary endpoint was the proportion of subjects with HIV RNA > 50 copies/mL at Week 48. No subjects had HIV RNA > 50 copies/mL. Ninety-one percent (78/86) of subjects remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 48. Eight subjects did not have virologic data at the Week 48 timepoint due to discontinuation or missing data.
Structured Label Content
Section 42229-5 (42229-5)
Clinical Trials in Adults with No Antiretroviral Treatment History
The primary safety assessment of BIKTARVY was based on data from two randomized, double-blind, active-controlled trials, Trial 1489 and Trial 1490, that enrolled 1274 HIV-1 infected adult subjects with no antiretroviral treatment history through Week 144. After Week 144, subjects received open-label BIKTARVY in an optional extension phase for an additional 96 weeks (end of study). A total of 634 and 1025 subjects received one tablet of BIKTARVY once daily during the double-blind (Week 144) and extension phases, respectively [see Clinical Studies (14.2)].
The most common adverse reactions (all Grades) reported in at least 5% of subjects in the BIKTARVY group in either Trial 1489 or Trial 1490 were diarrhea, nausea, and headache. The proportion of subjects who discontinued treatment through Week 144 with BIKTARVY, abacavir [ABC]/dolutegravir [DTG]/ lamivudine [3TC]), or DTG + FTC/TAF, due to adverse events, regardless of severity, was 1%, 2%, and 2%, respectively. Table 1 displays the frequency of adverse reactions (all Grades) greater than or equal to 2% in the BIKTARVY group.
| Trial 1489 | Trial 1490 | |||
|---|---|---|---|---|
| Adverse Reactions | BIKTARVY N=314 |
ABC/DTG/3TC N=315 |
BIKTARVY N=320 |
DTG + FTC/TAF N=325 |
| Diarrhea | 6% | 4% | 3% | 3% |
| Nausea | 6% | 18% | 3% | 5% |
| Headache | 5% | 5% | 4% | 3% |
| Fatigue | 3% | 4% | 2% | 2% |
| Abnormal dreams | 3% | 3% | <1% | 1% |
| Dizziness | 2% | 3% | 2% | 1% |
| Insomnia | 2% | 3% | 2% | <1% |
| Abdominal distention | 2% | 2% | 1% | 2% |
Additional adverse reactions (all Grades) occurring in less than 2% of subjects administered BIKTARVY in Trials 1489 and 1490 included vomiting, flatulence, dyspepsia, abdominal pain, rash, and depression.
Suicidal ideation, suicide attempt, and depression suicidal occurred in 2% of subjects administered BIKTARVY; these events occurred primarily in subjects with a preexisting history of depression, prior suicide attempt or psychiatric illness.
The majority (84%) of adverse events associated with BIKTARVY were Grade 1.
Adverse reactions in the open-label extension phases of Trials 1489 and 1490 were similar to those observed in subjects administered BIKTARVY in the Week 144 analysis.
Section 42230-3 (42230-3)
| Patient Information BIKTARVY® (bik-TAR-vee) (bictegravir, emtricitabine, and tenofovir alafenamide) tablets |
|
|---|---|
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised:02/2024 |
| Important: Ask your healthcare provider or pharmacist about medicines that should not be taken with BIKTARVY. For more information, see "What should I tell my healthcare provider before taking BIKTARVY?" | |
|
What is the most important information I should know about BIKTARVY? |
|
| BIKTARVY can cause serious side effects, including: | |
|
|
| For more information about side effects, see "What are the possible side effects of BIKTARVY?" | |
|
What is BIKTARVY?
BIKTARVY is a prescription medicine that is used without other human immunodeficiency virus-1 (HIV-1) medicines to treat HIV-1 infection in adults and children who weigh at least 31 pounds (14 kg): |
|
|
|
| HIV-1 is the virus that causes Acquired Immune Deficiency Syndrome (AIDS). BIKTARVY contains the medicines bictegravir, emtricitabine, and tenofovir alafenamide. It is not known if BIKTARVY is safe and effective in children who weigh less than 31 pounds (14 kg). |
|
| Do not take BIKTARVY if you also take a medicine that contains: | |
|
|
|
What should I tell my healthcare provider before taking BIKTARVY? |
|
Some medicines may interact with BIKTARVY. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
|
|
How should I take BIKTARVY?
|
|
|
What are the possible side effects of BIKTARVY? |
|
| BIKTARVY may cause serious side effects, including: | |
|
|
| The most common side effects of BIKTARVY are diarrhea, nausea, and headache. | |
| These are not all of the possible side effects of BIKTARVY. | |
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |
How should I store BIKTARVY?
|
|
| Keep BIKTARVY and all medicines out of reach of children. | |
| General information about the safe and effective use of BIKTARVY. | |
| Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use BIKTARVY for a condition for which it was not prescribed. Do not give BIKTARVY to other people, even if they have the same symptoms you have. It may harm them. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about BIKTARVY that is written for health professionals. | |
| What are the ingredients in BIKTARVY? | |
| Active ingredients: bictegravir, emtricitabine, and tenofovir alafenamide. | |
| Inactive ingredients: croscarmellose sodium, magnesium stearate, and microcrystalline cellulose. | |
| The tablets are film-coated with a coating material containing iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. | |
| Manufactured and distributed by: Gilead Sciences, Inc. Foster City, CA 94404 | |
| BIKTARVY is a trademark of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners. | |
| © 2024 Gilead Sciences, Inc. All rights reserved. 210251-GS-012 | |
| For more information, call 1-800-445-3235 or go to www.BIKTARVY.com. |
Section 43683-2 (43683-2)
| Indications and Usage (1) | 02/2024 |
Biktarvy (BIKTARVY)
10 Overdosage (10 OVERDOSAGE)
No data are available on overdose of BIKTARVY in patients. If overdose occurs, monitor the patient for evidence of toxicity. Treatment of overdose with BIKTARVY consists of general supportive measures including monitoring of vital signs as well as observation of the clinical status of the patient.
Hemodialysis treatment removes approximately 30% of the FTC dose over a 3-hour dialysis period starting within 1.5 hours of FTC dosing (blood flow rate of 400 mL/min and a dialysate flow rate of 600 mL/min). It is not known whether FTC can be removed by peritoneal dialysis.
Tenofovir is efficiently removed by hemodialysis with an extraction coefficient of approximately 54%.
11 Description (11 DESCRIPTION)
BIKTARVY (bictegravir, emtricitabine, and tenofovir alafenamide) is a fixed dose combination tablet containing bictegravir (BIC), emtricitabine (FTC), and tenofovir alafenamide (TAF) for oral administration.
- BIC is an integrase strand transfer inhibitor (INSTI).
- FTC, a synthetic nucleoside analog of cytidine, is an HIV nucleoside analog reverse transcriptase inhibitor (HIV NRTI).
- TAF, an HIV NRTI, is converted in vivo to tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate.
BIKTARVY tablets are available in two dose strengths:
- 50 mg/200 mg/25 mg tablet containing 50 mg of BIC (equivalent to 52.5 mg of bictegravir sodium),
200 mg of FTC, and 25 mg of TAF (equivalent to 28 mg of tenofovir alafenamide fumarate). - 30 mg/120 mg/15 mg tablet containing 30 mg of BIC (equivalent to 31.5 mg of bictegravir sodium), 120 mg of FTC, and 15 mg of TAF (equivalent to 16.8 mg of tenofovir alafenamide fumarate).
Both dose strengths of BIKTARVY tablets include the following inactive ingredients: croscarmellose sodium, magnesium stearate, and microcrystalline cellulose. The tablets for both dose strengths are film-coated with a coating material containing iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
8.4 Pediatric Use
The safety and effectiveness of BIKTARVY have been established as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in pediatric patients weighing at least 14 kg:
- who have no antiretroviral treatment history or
- to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no known or suspected resistance to bictegravir or tenofovir [see Indications and Usage (1), and Dosage and Administration (2.2, 2.3)].
Use of BIKTARVY in pediatric patients weighing at least 14 kg is supported by the following:
- trials in adults [see Clinical Studies (14.1)]
- an open-label trial in three age-based cohorts of virologically-suppressed pediatric subjects [see Clinical Studies (14.4)]
- Cohort 1: 12 to less than 18 years of age and weighing at least 35 kg receiving BIKTARVY through Week 48 (N=50),
- Cohort 2: 6 to less than 12 years of age and weighing at least 25 kg receiving BIKTARVY through Week 24 (N=50), and
- Cohort 3: at least 2 years of age and weighing at least 14 to less than 25 kg through Week 24 (N=22). No pediatric subjects 2 years of age were enrolled; of the 6 pediatric subjects who were 3 years of age at enrollment, 3 subjects weighed between 14 to less than 15 kg.
The safety and efficacy of BIKTARVY in these pediatric subjects were similar to that in adults, and there was no clinically significant change in exposure for the components of BIKTARVY [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.4)].
Safety and effectiveness of BIKTARVY in pediatric patients weighing less than 14 kg have not been established.
8.5 Geriatric Use
Clinical trials in virologically-suppressed subjects (Trials 4449, 1844, and 1878) included 111 subjects aged 65 years and over who received BIKTARVY, including 86 patients from an open-label, single-arm trial of subjects aged 65 years and over who were switched from their previous antiretroviral regimen to BIKTARVY [see Clinical Studies (14.3) ]. Of the total number of BIKTARVY-treated patients in these trials, 100 (90%) were 65 to 74 years of age, and 11 (10%) were 75 to 84 years of age. No overall differences in safety or effectiveness were observed between elderly subjects and adults between 18 and less than 65 years of age, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
4 Contraindications (4 CONTRAINDICATIONS)
BIKTARVY is contraindicated to be co-administered with:
- dofetilide due to the potential for increased dofetilide plasma concentrations and associated serious and/or life-threatening events [see Drug Interactions (7.5)].
- rifampin due to decreased BIC plasma concentrations, which may result in the loss of therapeutic effect and development of resistance to BIKTARVY [see Drug Interactions (7.5)].
6 Adverse Reactions (6 ADVERSE REACTIONS)
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbations of Hepatitis B [see Warnings and Precautions (5.1)].
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.3)].
- New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.4)].
- Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.5)].
7 Drug Interactions (7 DRUG INTERACTIONS)
8.6 Renal Impairment
The pharmacokinetics, safety, virologic and immunologic responses of FTC and TAF (components of BIKTARVY) were evaluated in a single arm, open-label trial (Trial 1825) in virologically-suppressed adults with ESRD (estimated creatinine clearance of less than 15 mL/min) on chronic hemodialysis treated with FTC+TAF in combination with elvitegravir and cobicistat as a fixed-dose combination tablet for 96 weeks (N=55). In an extension phase of Trial 1825, 10 virologically-suppressed subjects switched to BIKTARVY and all remained virologically suppressed for 48 weeks [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.3)].
No dosage adjustment of BIKTARVY is recommended in patients with estimated creatinine clearance greater than or equal to 30 mL/min, or in virologically-suppressed adults (estimated creatinine clearance below 15 mL/min) who are receiving chronic hemodialysis. On days of hemodialysis, administer the daily dose of BIKTARVY after completion of hemodialysis treatment [see Dosage and Administration (2.2)]
BIKTARVY is not recommended in patients with estimated creatinine clearance of below 30 mL/min, by Cockcroft-Gault, or patients with ESRD (estimated creatinine clearance below 15 mL/min) who are not receiving chronic dialysis, or patients with no antiretroviral treatment history and ESRD who are receiving chronic dialysis, as the safety and/or efficacy of BIKTARVY has not been established in these populations [see Dosage and Administration (2.4), Warnings and Precautions (5.4), and Clinical Pharmacology (12.3)].
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of BIKTARVY components are provided in Table 4. The multiple dose PK parameters of BIKTARVY components (based on population pharmacokinetic analysis) are provided in Table 5.
| Bictegravir (BIC) | Emtricitabine (FTC) | Tenofovir Alafenamide (TAF) | ||
|---|---|---|---|---|
| PBMCs=peripheral blood mononuclear cells; CES1=carboxylesterase 1 | ||||
| Absorption | ||||
| Tmax (h) Values reflect administration of BIKTARVY with or without food.
|
2.0–4.0 | 1.5–2.0 | 0.5–2.0 | |
| Effect of high-fat meal (relative to fasting) Values refer to geometric mean ratio [high-fat meal/ fasting] in PK parameters and (90% confidence interval). High fat meal is approximately 800 kcal, 50% fat.
|
AUC ratio | 1.24 (1.16, 1.33) | 0.96 (0.93, 0.99) | 1.63 (1.43, 1.85) |
| Cmax ratio | 1.13 (1.06, 1.20) | 0.86 (0.78, 0.93) | 0.92 (0.73, 1.14) | |
| Distribution | ||||
| % bound to human plasma proteins | >99 | <4 | ~80 | |
| Blood-to-plasma ratio | 0.64 | 0.6 | 1.0 | |
| Elimination | ||||
| t1/2 (h) t1/2 values refer to median (Q1, Q3) terminal plasma half-life. Note that the active metabolite of TAF, tenofovir diphosphate, has a half-life of 150–180 hours within PBMCs.
|
17.3 (14.8, 20.7) | 10.4 (9.0, 12.0) | 0.51 (0.45, 0.62) | |
| Metabolism | ||||
| Metabolic pathway(s) | CYP3A UGT1A1 |
Not significantly metabolized | Cathepsin A
In vivo, TAF is hydrolyzed within cells to form tenofovir (major metabolite), which is phosphorylated to the active metabolite, tenofovir diphosphate. In vitro studies have shown that TAF is metabolized to tenofovir by cathepsin A in PBMCs and macrophages; and by CES1 in hepatocytes. (PBMCs)CES1 (hepatocytes) |
|
| Excretion | ||||
| Major route of elimination | Metabolism | Glomerular filtration and active tubular secretion | Metabolism | |
| % of dose excreted in urine Dosing in mass balance studies: single dose administration of [14C] BIC; single dose administration of [14C] FTC after multiple dosing of FTC for ten days; single dose administration of [14C] TAF.
|
35 | 70 | <1 | |
| % of dose excreted in feces | 60.3 | 13.7 | 31.7 |
| Parameter Mean (CV%) | Bictegravir | Emtricitabine | Tenofovir Alafenamide |
|---|---|---|---|
| CV=Coefficient of Variation; NA=Not Applicable | |||
| Cmax
(microgram per mL) |
6.15 (22.9) | 2.13 (34.7) | 0.121 (15.4) |
| AUCtau
(microgram∙h per mL) |
102 (26.9) | 12.3 (29.2) | 0.142 (17.3) |
| Ctrough
(microgram per mL) |
2.61 (35.2) | 0.096 (37.4) | NA |
8.7 Hepatic Impairment
No dosage adjustment of BIKTARVY is recommended in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. BIKTARVY has not been studied in patients with severe hepatic impairment (Child-Pugh Class C). Therefore, BIKTARVY is not recommended for use in patients with severe hepatic impairment [see Dosage and Administration (2.4), and Clinical Pharmacology (12.3)].
1 Indications and Usage (1 INDICATIONS AND USAGE)
BIKTARVY is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 14 kg:
- who have no antiretroviral treatment history or
- to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no known or suspected substitutions associated with resistance to bictegravir or tenofovir.
12.1 Mechanism of Action
BIKTARVY is a fixed dose combination of antiretroviral drugs bictegravir (BIC), emtricitabine (FTC), and tenofovir alafenamide (TAF) [see Microbiology (12.4)].
5 Warnings and Precautions (5 WARNINGS AND PRECAUTIONS)
- Immune reconstitution syndrome: May necessitate further evaluation and treatment. (5.3)
- New onset or worsening renal impairment: Assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein when initiating BIKTARVY and during therapy as clinically appropriate in all patients. Also assess serum phosphorus in patients with chronic kidney disease. (5.4)
- Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. (5.5)
2 Dosage and Administration (2 DOSAGE AND ADMINISTRATION)
- Testing: Prior to or when initiating BIKTARVY test for hepatitis B virus infection. Prior to or when initiating BIKTARVY, and during treatment, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus. (2.1)
- Recommended dosage in adults and pediatric patients weighing at least 25 kg: One tablet containing 50 mg BIC, 200 mg FTC, and 25 mg TAF taken once daily with or without food. (2.2)
- Recommended dosage in pediatric patients weighing at least 14 kg to less than 25 kg: One tablet containing 30 mg BIC, 120 mg FTC, and 15 mg TAF taken once daily with or without food. (2.3)
- Renal impairment: BIKTARVY is not recommended in patients with estimated creatinine clearance of 15 to below 30 mL/min, or below 15 mL/min who are not receiving chronic hemodialysis, or below 15 mL/min who have no antiretroviral treatment history. (2.4)
- Hepatic impairment: BIKTARVY is not recommended in patients with severe hepatic impairment. (2.5)
3 Dosage Forms and Strengths (3 DOSAGE FORMS AND STRENGTHS)
BIKTARVY tablets are available in two dose strengths:
- 50 mg/200 mg/25 mg tablets: 50 mg of bictegravir (BIC) (equivalent to 52.5 mg of bictegravir sodium), 200 mg of emtricitabine (FTC), and 25 mg of tenofovir alafenamide (TAF) (equivalent to 28 mg of tenofovir alafenamide fumarate). These tablets are purplish brown, capsule-shaped, film-coated, and debossed with "GSI" on one side and "9883" on the other side.
- 30 mg/120 mg/15 mg tablets: 30 mg of BIC (equivalent to 31.5 mg of bictegravir sodium), 120 mg of FTC, and 15 mg of TAF (equivalent to 16.8 mg of tenofovir alafenamide fumarate). These tablets are pink, capsule-shaped, film-coated, and debossed with "GSI" on one side and "B" on the other side.
6.2 Postmarketing Experience
The following events have been identified during post approval use of BIKTARVY or products containing TAF. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
8 Use in Specific Populations (8 USE IN SPECIFIC POPULATIONS)
- Pediatrics: Not recommended for patients weighing less than 14 kg. (8.4)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
17 Patient Counseling Information (17 PATIENT COUNSELING INFORMATION)
Advise the patient to read the FDA-approved patient labeling (Patient Information).
5.3 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections [such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia (PCP), or tuberculosis], which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
7.4 Drugs Affecting Renal Function
Because FTC and tenofovir are primarily excreted by the kidneys by a combination of glomerular filtration and active tubular secretion, coadministration of BIKTARVY with drugs that reduce renal function or compete for active tubular secretion may increase concentrations of FTC, tenofovir, and other renally eliminated drugs and this may increase the risk of adverse reactions. Some examples of drugs that are eliminated by active tubular secretion include, but are not limited to, acyclovir, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.4)].
14.1 Description of Clinical Trials
The efficacy and safety of BIKTARVY were evaluated in the trials summarized in Table 12.
| Trial | Population | Trial Arms (N) | Timepoint (Week) |
|---|---|---|---|
| OLE = open-label extension | |||
| Trial 1489 Randomized, double blind, active controlled trial.
(NCT 02607930) |
Adults with no antiretroviral treatment history | BIKTARVY (314) ABC/DTG/3TC (315) |
144 + 96 (OLE) 144-week double-blind active controlled phase followed by an extension phase in which 1025 subjects from Trials 1489 and 1490 received open-label BIKTARVY for 96 weeks.
|
| Trial 1490
(NCT 02607956) |
BIKTARVY (320) DTG + FTC/TAF(325) |
144 + 96 (OLE) | |
| Trial 1844
(NCT 02603120) |
Virologically-suppressed HIV-1 RNA less than 50 copies per mL. adults |
BIKTARVY (282) ABC/DTG/3TC (281) |
48 |
| Trial 1878 Randomized, open-label, active controlled trial.
(NCT 02603107) |
BIKTARVY (290) ATV or DRV (with cobicistat or ritonavir) plus either FTC/TDF or ABC/3TC (287) |
48 | |
| Trial 4030
(NCT 03110380) |
BIKTARVY (284 [47 with M184V/I]) DTG plus FTC/TAF (281 [34 with M184V/I]) |
48 | |
| Trial 1825 Open-label trial.
(NCT 02600819) |
Virologically-suppressed adults with ESRD End stage renal disease (estimated creatinine clearance of less than 15 mL/min by Cockcroft-Gault method). receiving chronic hemodialysis |
FTC+TAF in combination with elvitegravir and cobicistat as a fixed-dose combination (55). In an extension phase of Trial 1825, 10 virologically-suppressed subjects switched to BIKTARVY. | 48 Subjects received FTC+TAF in combination with elvitegravir and cobicistat for 96 weeks, followed by an extension phase in which 10 subjects received BIKTARVY for 48 weeks.
|
| Trial 4449
(NCT 03405935) |
Virologically-suppressed adults aged 65 years and over | BIKTARVY (86) | 48 |
| Trial 1474
(cohort 1) (NCT 02881320) |
Virologically-suppressed adolescents between the ages of 12 to less than 18 years (at least 35 kg) | BIKTARVY (50) | 48 |
| Trial 1474
(cohort 2) (NCT 02881320) |
Virologically-suppressed children between the ages of 6 to less than 12 years (at least 25 kg) | BIKTARVY (50) | 24 |
| Trial 1474
(cohort 3) (NCT 02881320) |
Virologically-suppressed children at least 2 years of age (at least 14 to less than 25 kg) | BIKTARVY (22) | 24 |
16 How Supplied/storage and Handling (16 HOW SUPPLIED/STORAGE AND HANDLING)
Product: 50090-6247
NDC: 50090-6247-0 30 TABLET in a BOTTLE, PLASTIC
7.1 Other Antiretroviral Medications
Because BIKTARVY is a complete regimen, coadministration with other antiretroviral medications for the treatment of HIV-1 infection is not recommended [see Indications and Usage (1)]. Comprehensive information regarding potential drug-drug interactions with other antiretroviral medications is not provided because the safety and efficacy of concomitant HIV-1 antiretroviral therapy is unknown.
13.2 Animal Toxicology And/or Pharmacology (13.2 Animal Toxicology and/or Pharmacology)
Minimal to slight infiltration of mononuclear cells in the posterior uvea was observed in dogs with similar severity after three and nine month administration of TAF; reversibility was seen after a three month recovery period. No eye toxicity was observed in the dog at systemic exposures of 7 (TAF) and 14 (tenofovir) times the exposure seen in humans with the recommended daily dose of BIKTARVY.
5.4 New Onset Or Worsening Renal Impairment (5.4 New Onset or Worsening Renal Impairment)
Postmarketing cases of renal impairment, including acute renal failure, proximal renal tubulopathy (PRT), and Fanconi syndrome, have been reported with TAF-containing products; while most of these cases were characterized by potential confounders that may have contributed to the reported renal events, it is also possible these factors may have predisposed patients to tenofovir-related adverse events [see Adverse Reactions (6.1, 6.2)]. BIKTARVY is not recommended in patients with severe renal impairment (estimated creatinine clearance of 15 to below 30 mL/min), or patients with ESRD (estimated creatinine clearance below 15 mL/min) who are not receiving chronic hemodialysis, or patients with no antiretroviral treatment history and ESRD who are receiving chronic hemodialysis [see Dosage and Administration (2.4), and Use in Specific Populations (8.6)].
Patients taking tenofovir prodrugs who have impaired renal function and those taking nephrotoxic agents including non-steroidal anti-inflammatory drugs are at increased risk of developing renal-related adverse reactions.
Prior to or when initiating BIKTARVY, and during treatment with BIKTARVY, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus. Discontinue BIKTARVY in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome.
7.2 Potential for Biktarvy to Affect Other Drugs (7.2 Potential for BIKTARVY to Affect Other Drugs)
BIC inhibits organic cation transporter 2 (OCT2) and multidrug and toxin extrusion transporter 1 (MATE1) in vitro. Coadministration of BIKTARVY with drugs that are substrates of OCT2 and MATE1 (e.g., dofetilide) may increase their plasma concentrations (see Table 3).
5.5 Lactic Acidosis/severe Hepatomegaly With Steatosis (5.5 Lactic Acidosis/Severe Hepatomegaly with Steatosis)
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including emtricitabine, a component of BIKTARVY, and tenofovir DF, another prodrug of tenofovir, alone or in combination with other antiretrovirals. Treatment with BIKTARVY should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
Warning: Post Treatment Acute Exacerbation of Hepatitis B (WARNING: POST TREATMENT ACUTE EXACERBATION OF HEPATITIS B)
Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY.
Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue BIKTARVY. If appropriate, anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)].
14.4 Clinical Trial Results in Pediatric Subjects With Hiv 1 (14.4 Clinical Trial Results in Pediatric Subjects with HIV-1)
In Trial 1474, an open-label, single arm trial the efficacy, safety, and pharmacokinetics of BIKTARVY in HIV-1 infected pediatric subjects were evaluated in virologically-suppressed adolescents between the ages of 12 to less than 18 years weighing at least 35 kg (N=50), in virologically-suppressed children between the ages of 6 to less than 12 years weighing at least 25 kg (N=50), and in virologically-suppressed children at least 2 years of age and weighing at least 14 to less than 25 kg (N=22).
2.4 Not Recommended in Patients With Severe Renal Impairment (2.4 Not Recommended in Patients with Severe Renal Impairment)
BIKTARVY is not recommended in patients with [see Dosage and Administration (2.2, 2.3), and Use in Specific Populations (8.6)]:
- severe renal impairment (estimated creatinine clearance of 15 to below 30 mL/min); or
- end stage renal disease (ESRD; estimated creatinine clearance below 15 mL/min who are not receiving chronic hemodialysis; or
- no antiretroviral treatment history and ESRD who are receiving chronic hemodialysis.
7.5 Established and Potentially Significant Drug Interactions
Table 3 provides a listing of established or potentially clinically significant drug interactions with recommended prevention or management strategies. The drug interactions described are based on studies conducted with either BIKTARVY, the components of BIKTARVY (BIC, FTC, and TAF) as individual agents, or are drug interactions that may occur with BIKTARVY [see Contraindications (4), Warnings and Precautions (5.2), and Clinical Pharmacology (12.3)].
| Concomitant Drug Class: Drug Name | Effect on Concentration ↑ = Increase, ↓ = Decrease.
|
Clinical Comment |
|---|---|---|
|
Antiarrhythmics:
dofetilide |
↑ Dofetilide | Coadministration is contraindicated due to the potential for serious and/or life-threatening events associated with dofetilide therapy [see Contraindications (4)]. |
|
Anticonvulsants:
carbamazepine Drug-drug interaction study was conducted with either BIKTARVY or its components as individual agents.
oxcarbazepine phenobarbital phenytoin |
↓ BIC ↓ TAF |
Coadministration with alternative anticonvulsants should be considered. |
|
Antimycobacterials:
rifabutin rifampin , Strong inducer of CYP3Aand P-gp, and inducer of UGT1A1.
rifapentine |
↓ BIC ↓ TAF |
Coadministration with rifampin is contraindicated due to the effect of rifampin on the BIC component of BIKTARVY [see Contraindications (4)]. Coadministration with rifabutin or rifapentine is not recommended. |
|
Herbal Products:
St. John's wort The induction potency of St. John's wort may vary widely based on preparation.
|
↓ BIC ↓ TAF |
Coadministration with St. John's wort is not recommended. |
|
Medications or oral supplements containing polyvalent cations (e.g., Mg, Al, Ca, Fe):
Calcium or iron supplements Cation-containing antacids or laxatives Sucralfate Buffered medications |
↓ BIC |
Antacids containing Al/Mg:
BIKTARVY can be taken at least 2 hours before or 6 hours after taking antacids containing Al/Mg. Routine administration of BIKTARVY together with, or 2 hours after, antacids containing Al/Mg is not recommended. Supplements or Antacids containing Calcium or Iron: BIKTARVY and supplements or antacids containing calcium or iron can be taken together with food. Routine administration of BIKTARVY under fasting conditions together with, or 2 hours after, supplements or antacids containing calcium or iron is not recommended. |
| Metformin | ↑ Metformin | Refer to the prescribing information of metformin for assessing the benefit and risk of concomitant use of BIKTARVY and metformin. |
2.1 Testing When Initiating and During Treatment With Biktarvy (2.1 Testing When Initiating and During Treatment with BIKTARVY)
Prior to or when initiating BIKTARVY, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to or when initiating BIKTARVY, and during treatment with BIKTARVY, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus [see Warnings and Precautions (5.4)].
2.5 Not Recommended in Patients With Severe Hepatic Impairment (2.5 Not Recommended in Patients with Severe Hepatic Impairment)
BIKTARVY is not recommended in patients with severe hepatic impairment (Child-Pugh Class C) [see Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
7.6 Drugs Without Clinically Significant Interactions With Biktarvy (7.6 Drugs without Clinically Significant Interactions with BIKTARVY)
Based on drug interaction studies conducted with BIKTARVY or the components of BIKTARVY, no clinically significant drug interactions have been observed when BIKTARVY is combined with the following drugs: ethinyl estradiol, ledipasvir/sofosbuvir, midazolam, norgestimate, sertraline, sofosbuvir, sofosbuvir/velpatasvir, and sofosbuvir/velpatasvir/voxilaprevir.
7.3 Potential Effect of Other Drugs On One Or More Components of Biktarvy (7.3 Potential Effect of Other Drugs on One or More Components of BIKTARVY)
BIC is a substrate of CYP3A and UGT1A1. A drug that is a strong inducer of CYP3A and also an inducer of UGT1A1 can substantially decrease the plasma concentrations of BIC which may lead to loss of therapeutic effect of BIKTARVY and development of resistance [see Clinical Pharmacology (12.3)].
The use of BIKTARVY with a drug that is a strong inhibitor of CYP3A and also an inhibitor of UGT1A1 may significantly increase the plasma concentrations of BIC.
TAF is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Co-administration of drugs that inhibit P-gp and BCRP may increase the absorption and plasma concentrations of TAF [see Clinical Pharmacology (12.3)]. Co-administration of drugs that induce P-gp activity are expected to decrease the absorption of TAF, resulting in decreased plasma concentration of TAF, which may lead to loss of therapeutic effect of BIKTARVY and development of resistance (see Table 3).
2.2 Recommended Dosage in Adults and Pediatric Patients Weighing At Least 25 Kg (2.2 Recommended Dosage in Adults and Pediatric Patients Weighing at Least 25 kg)
BIKTARVY is a three-drug fixed dose combination product containing bictegravir (BIC), emtricitabine (FTC), and tenofovir alafenamide (TAF). The recommended dosage of BIKTARVY is one tablet containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF taken orally once daily with or without food in:
- adults and pediatric patients weighing at least 25 kg with an estimated creatinine clearance greater than or equal to 30 mL/min; or
- virologically-suppressed adults with an estimated creatinine clearance below 15 mL/min who are receiving chronic hemodialysis. On days of hemodialysis, administer the daily dose of BIKTARVY after completion of hemodialysis treatment [see Use in Specific Populations (8.4, 8.6), and Clinical Pharmacology (12.3)].
5.2 Risk of Adverse Reactions Or Loss of Virologic Response Due to Drug Interactions (5.2 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions)
The concomitant use of BIKTARVY with certain other drugs may result in known or potentially significant drug interactions, some of which may lead to [see Contraindications (4), and Drug Interactions (7.5)]:
- Loss of therapeutic effect of BIKTARVY and possible development of resistance.
- Possible clinically significant adverse reactions from greater exposures of concomitant drugs.
See Table 3 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during BIKTARVY therapy; review concomitant medications during BIKTARVY therapy; and monitor for the adverse reactions associated with the concomitant drugs.
5.1 Severe Acute Exacerbation of Hepatitis B in Patients Coinfected With Hiv 1 and Hbv (5.1 Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV)
Patients with HIV-1 should be tested for the presence of chronic hepatitis B virus (HBV) infection before or when initiating antiretroviral therapy [see Dosage and Administration (2.1)].
Severe acute exacerbations of hepatitis B (e.g., liver decompensation and liver failure) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing FTC and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY. Patients coinfected with HIV-1 and HBV who discontinue BIKTARVY should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since post-treatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
2.3 Recommended Dosage in Pediatric Patients Weighing At Least 14 Kg to Less Than 25 Kg (2.3 Recommended Dosage in Pediatric Patients Weighing at Least 14 kg to Less than 25 kg)
The recommended dosage of BIKTARVY is one tablet containing 30 mg of BIC, 120 mg of FTC, and 15 mg of TAF taken orally once daily with or without food in:
- pediatric patients weighing at least 14 kg to less than 25 kg with an estimated creatinine clearance greater than or equal to 30 mL/min [see Use in Specific Populations (8.4, 8.6), and Clinical Pharmacology (12.3)].
For children unable to swallow a whole tablet, the tablet can be split and each part taken separately as long as all parts are ingested within approximately 10 minutes.
14.2 Clinical Trial Results in Adults With Hiv 1 and No Antiretroviral Treatment History (14.2 Clinical Trial Results in Adults with HIV-1 and No Antiretroviral Treatment History)
In Trial 1489, adults were randomized in a 1:1 ratio to receive either BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) (N=314) or ABC/DTG/3TC (600 mg/50 mg/300 mg) (N=315) once daily. In Trial 1490, subjects were randomized in a 1:1 ratio to receive either BIKTARVY (N=320) or DTG + FTC/TAF (50 mg + 200 mg/25 mg) (N=325) once daily.
In Trial 1489, the mean age was 34 years (range 18–71), 90% were male, 57% were White, 36% were Black, and 3% were Asian. 22% of patients identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.4 log10 copies/mL (range 1.3–6.5). The mean baseline CD4+ cell count was 464 cells per mm3 (range 0–1424) and 11% had CD4+ cell counts less than 200 cells per mm3. 16% of subjects had baseline viral loads greater than 100,000 copies per mL.
In Trial 1490, the mean age was 37 years (range 18–77), 88% were male, 59% were White, 31% were Black, and 3% were Asian. 25% of patients identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.4 log10 copies/mL (range 2.3–6.6). The mean baseline CD4+ cell count was 456 cells per mm3 (range 2–1636) and 12% had CD4+ cell counts less than 200 cells per mm3. 19% of subjects had baseline viral loads greater than 100,000 copies per mL.
In both trials, subjects were stratified by baseline HIV-1 RNA (less than or equal to 100,000 copies per mL, greater than 100,000 copies per mL to less than or equal to 400,000 copies per mL, or greater than 400,000 copies per mL), by CD4 count (less than 50 cells per mm3, 50–199 cells per mm3, or greater than or equal to 200 cells per mm3), and by region (US or ex-US).
Treatment outcomes of Trials 1489 and 1490 through Week 144 are presented in Table 13.
| Trial 1489 | Trial 1490 | |||
|---|---|---|---|---|
| BIKTARVY (N=314) |
ABC/DTG/3TC (N=315) |
BIKTARVY (N=320) |
DTG + FTC/TAF (N=325) |
|
| HIV-1 RNA < 50 copies/mL | 82% | 84% | 82% | 84% |
| Treatment Difference (95% CI) BIKTARVY vs. Comparator | -2.6% (-8.5% to 3.4%) | -1.9% (-7.8% to 3.9%) | ||
|
HIV-1 RNA ≥ 50 copies/mL Includes subjects who had ≥ 50 copies/mL in the Week 144 window; subjects who discontinued early due to lack or loss of efficacy; subjects who discontinued for reasons other than an adverse event (AE), death or lack or loss of efficacy and at the time of discontinuation had a viral value of ≥ 50 copies/mL.
|
1% | 3% | 5% | 3% |
| No Virologic Data at Week 144 Window | 18% | 13% | 13% | 13% |
| Discontinued Study Drug Due to AE or Death Includes subjects who discontinued due to AE or death at any time point from Day 1 through the time window if this resulted in no virologic data on treatment during the specified window.
|
1% | 2% | 3% | 3% |
| Discontinued Study Drug Due to Other Reasons and Last Available HIV-1 RNA <50 copies/mL Includes subjects who discontinued for reasons other than an AE, death, or lack or loss of efficacy, e.g., withdrew consent, loss to follow-up, etc.
|
16% | 11% | 11% | 9% |
| Missing Data During Window but on Study Drug | 1% | <1% | 0% | 1% |
Treatment outcomes were similar across subgroups by age, sex, race, baseline viral load, and baseline CD4+ cell count.
In Trials 1489 and 1490, the mean increase from baseline in CD4+ count at Week 144 was 299 and 317 cells per mm3 in the BIKTARVY and ABC/DTG/3TC groups, respectively, and 278 and 289 cells per mm3 in the BIKTARVY and DTG + FTC/TAF groups, respectively.
14.3 Clinical Trial Results in Adults With Virologically Suppressed Hiv 1 Who Switched to Biktarvy (14.3 Clinical Trial Results in Adults with Virologically-Suppressed HIV-1 Who Switched to BIKTARVY)
In Trial 1844, the efficacy and safety of switching from a regimen of DTG + ABC/3TC or ABC/DTG/3TC to BIKTARVY were evaluated in a randomized, double-blind trial of virologically-suppressed (HIV-1 RNA less than 50 copies per mL) HIV-1 infected adults (N=563, randomized and dosed). Subjects must have been stably suppressed (HIV-1 RNA less than 50 copies per mL) on their baseline regimen for at least 3 months prior to trial entry and had no history of treatment failure. Subjects were randomized in a 1:1 ratio to either switch to BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) at baseline (N=282), or stay on their baseline antiretroviral regimen (N=281). Subjects had a mean age of 45 years (range 20–71), 89% were male, 73% were White, and 22% were Black. 17% of subjects identified as Hispanic/Latino. The mean baseline CD4+ cell count was 723 cells per mm3 (range 124–2444).
In Trial 1878, the efficacy and safety of switching from either ABC/3TC or FTC/TDF (200/300 mg) plus ATV or DRV (given with either cobicistat or ritonavir) to BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) were evaluated in a randomized, open-label study of virologically-suppressed HIV-1 infected adults (N=577, randomized and dosed). Subjects must have been stably suppressed on their baseline regimen for at least 6 months, must not have been previously treated with any INSTI, and had no history of treatment failure. Subjects were randomized in a 1:1 ratio to either switch to BIKTARVY (N=290) or stay on their baseline antiretroviral regimen (N=287). Subjects had a mean age of 46 years (range 20–79), 83% were male, 66% were White, and 26% were Black. 19% of subjects identified as Hispanic/Latino. The mean baseline CD4+ cell count was 663 cells per mm3 (range 62–2582). Subjects were stratified by prior treatment regimen. At screening, 15% of subjects were receiving ABC/3TC plus ATV or DRV (given with either cobicistat or ritonavir) and 85% of subjects were receiving FTC/TDF plus ATV or DRV (given with either cobicistat or ritonavir).
Treatment outcomes of Trials 1844 and 1878 through Week 48 are presented in Table 14.
| Trial 1844 | Trial 1878 | |||
|---|---|---|---|---|
| BIKTARVY (N=282) |
ABC/DTG/3TC (N=281) |
BIKTARVY (N=290) |
ATV- or DRV-based regimen ATV given with cobicistat or ritonavir or DRV given with cobicistat or ritonavir plus either FTC/TDF or ABC/3TC.
(N=287) |
|
|
HIV-1 RNA ≥ 50 copies/mL Includes subjects who had ≥ 50 copies/mL in the Week 48 window; subjects who discontinued early due to lack or loss of efficacy; subjects who discontinued for reasons other than lack or loss of efficacy and at the time of discontinuation had a viral value of ≥ 50 copies/mL.
|
1% | <1% | 2% | 2% |
| Treatment Difference (95% CI) | 0.7% (-1.0% to 2.8%) | 0.0% (-2.5% to 2.5%) | ||
| HIV-1 RNA < 50 copies/mL | 94% | 95% | 92% | 89% |
| No Virologic Data at Week 48 Window | 5% | 5% | 6% | 9% |
| Discontinued Study Drug Due to AE or Death and Last Available HIV-1 RNA < 50 copies/mL | 2% | 1% | 1% | 1% |
| Discontinued Study Drug Due to Other Reasons and Last Available HIV-1 RNA < 50 copies/mL Includes subjects who discontinued for reasons other than an AE, death, or lack or loss of efficacy, e.g., withdrew consent, loss to follow-up, etc.
|
2% | 3% | 3% | 7% |
| Missing Data During Window but on Study Drug | 2% | 1% | 2% | 2% |
In Trial 1844, treatment outcomes between treatment groups were similar across subgroups by age, sex, race, and region. The mean change from baseline in CD4+ count at Week 48 was -31 cells per mm3 in subjects who switched to BIKTARVY and 4 cells per mm3 in subjects who stayed on ABC/DTG/3TC.
In Trial 1878, treatment outcomes between treatment groups were similar across subgroups by age, sex, race, and region. The mean change from baseline in CD4+ count at Week 48 was 25 cells per mm3 in patients who switched to BIKTARVY and 0 cells per mm3 in patients who stayed on their baseline regimen.
In Trial 4030, the efficacy and safety of switching from DTG plus either FTC/TAF or FTC/TDF to BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) were evaluated in a randomized, double-blind study of virologically suppressed HIV-1 infected adults. Subjects must have been stably suppressed (HIV-1 RNA less than 50 copies/mL) on their baseline regimen for at least 6 months (if documented or suspected NRTI resistance), or at least 3 months (if no documented or suspected NRTI resistance) prior to trial entry. Subjects were randomized to switch to BIKTARVY (N=284) or to continue their prior treatment regimen, DTG+ F/TAF (N=281). The primary endpoint was the proportion of subjects with HIV-1 RNA ≥ 50 copies/mL at Week 48. At Week 48 the proportion of subjects with HIV-1 RNA ≥50 copies/mL was 0.4% (1/284) in the BIKTARVY group and 1.1% (3/281) in the DTG+F/TAF group (difference -0.7% [95%CI: -2.8%, 1.0%]).
Of the subjects receiving BIKTARVY, 47 had HIV-1 with pre-existing M184V or I resistance substitutions (M184M/V, M184M/I, M184V/I, M184V) in HIV-1 RT. Eighty-nine percent (42/47) of subjects with M184V or I remained suppressed (HIV-1 RNA < 50 copies/mL) and 11% (5/47 subjects) did not have virologic data at the Week 48 timepoint due to study drug discontinuation.
In Trial 1825, an open-label single arm trial, the efficacy, safety, and pharmacokinetics of FTC and TAF (components of BIKTARVY) were evaluated in virologically-suppressed adults with ESRD (estimated creatinine clearance of less than 15 mL/min) on chronic hemodialysis treated with FTC+TAF in combination with elvitegravir and cobicistat as a fixed-dose combination tablet for 96 weeks (N=55). In an extension phase of Trial 1825, 10 virologically-suppressed subjects switched to BIKTARVY and all subjects remained virologically suppressed (HIV-1 RNA < 50 copies/mL) for 48 weeks.
In Trial 4449, the efficacy and safety of switching from a stable antiretroviral regimen to BIKTARVY (containing 50 mg of BIC, 200 mg of FTC, and 25 mg of TAF) were evaluated in an open-label, single arm trial of virologically-suppressed (HIV-1 RNA less than 50 copies per mL) HIV-1 infected adults aged 65 years and over (N=86). Subjects treated with BIKTARVY had a mean age of 70 years (range: 65 to 80). The primary endpoint was the proportion of subjects with HIV RNA > 50 copies/mL at Week 48. No subjects had HIV RNA > 50 copies/mL. Ninety-one percent (78/86) of subjects remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 48. Eight subjects did not have virologic data at the Week 48 timepoint due to discontinuation or missing data.
Advanced Ingredient Data
Raw Label Data
All Sections (JSON)
Additional Information
Back to search View SPL set listing Open on DailyMed ↗
Source: dailymed · Ingested: 2026-02-15T11:40:53.903998 · Updated: 2026-03-14T22:04:26.530663